The Possible Role of Β-Cell Senescence in the Development of Type 2 Diabetes Mellitus
Keywords
Abstract
Introduction
Epidemiological studies of diabetes mellitus are regularly conducted by the World Health Organization (WHO) and the International Diabetes Federation (IDF). In 1998, investigators predicted that the number of people with type 1 and type 2 diabetes would increase from 135 million in 1995 to 300 million by 2025 [1]. At the end of 2019, IDF data were published, indicating that 453 million people had type 1 and type 2 diabetes in the world in 2019; according to forecasts, this number will reach 700 million in 2045. Indeed, all researchers have noted the trend of annual increases in diagnosed diabetes mellitus cases. Importantly, not all diabetes cases are diagnosed. Diabetes-related death statistics are especially dramatic. According to IDF data, among all people worldwide who died in the age range of 20 to 79 years in 2019, 4.2 million died of diabetes, i.e., one in 12 deaths from all diseases was associated with diabetes [2]. More than one-third of the world’s population is overweight or obese and therefore at risk of developing type 2 diabetes. To mitigate the effects of this pandemic, safer and more effective therapeutics are urgently needed. This resolution requires the use of animal models to discover, validate and optimize new therapeutics for safe use in humans [3]. Type 2 diabetes mellitus, characterized by insulin resistance, hyperglycemia, and hyperlipidemia, is known to be a complex metabolic disorder [4]. Insulin release from pancreatic β-cells is essential for maintaining normal glucose homeostasis in humans and animals. Abnormal insulin secretion underlies all forms of diabetes mellitus, a disease that is now reaching epidemic proportions throughout the world. Since insulin-producing pancreatic β-cells are the main target in both type 1 and type 2 diabetes mellitus [5], research is being conducted around the world to find approaches to reduce the destruction of pancreatic β-cells or even to stimulate their differentiation [6]. There are known markers for type 1 diabetes (for example, the presence of pancreatic autoantibodies in the blood), but the diagnosis of type 2 diabetes (T2D), which, like type 1 diabetes (T1D), is characterized by hyperglycemia, is still very difficult, and the exclusion of a T1D diagnosis is almost always used. The most significant difference between these two forms of diabetes is that T1D is characterized by the destruction of pancreatic β-cells, while T2D is characterized by a decrease in their functional activity [7], expressed as a loss of their secretory function and a decrease in the mass of β-cells [8]. Hyperglycemia leads to the production of reactive oxygen species (ROS) and reduces the effectiveness of the endogenous antioxidant defense system in diabetes mellitus [9]. Antioxidant defense mechanisms include both enzymatic and nonenzymatic systems. There is strong evidence for oxidative stress both in type 1 and type 2 diabetes [10-12]. At the same time, an increase in the production of reactive oxygen or nitrogen species increases the consumption of endogenous antioxidant enzymes (superoxide dismutase, glutathione peroxidase and catalase) and reduces the concentration of some low-molecular-weight antioxidants, such as vitamin D [13]. A decrease in the activity of peroxiredoxins under conditions of oxidative and carbonyl stress may be an important factor triggering the molecular mechanisms of damage to the vascular wall in atherosclerosis and diabetes mellitus [14]. The particular combinations of β-cell defects, ultimately inducing T2D, may differ between individuals and include variations in the development of β-cell mass, β-cell expansion, responses to ER stress and oxidative stress, insulin production and secretion, and intracellular changes in signaling pathways. Because some β-cells survive in the toxic environment in T2D but remain in an altered state of differentiation, strategies to revive these cells and rejuvenate their functions are needed [7]. We believe that it is necessary to identify mechanisms that stimulate β-cell proliferation or promote β-cell survival and use them to develop new therapeutic agents for the treatment of T2D. An important aspect of the development of T2D is aging and excess weight (obesity). Indeed, T2D may be considered an age-related disease [15]. Although changes in aged β-cell function and proliferation should affect insulin secretion, the contribution of β-cell senescence remains unclear. Insulin resistance has been found to accelerate β-cell aging, leading to loss of function and cellular identity and a worsening metabolic profile. Senolysis (removal of senescent cells) using senolytics was shown to improve glucose metabolism and β-cell function while reducing the expression of aging markers [16]. Thus, type 2 diabetes is an age-related disease characterized by a decrease in the mass and functional activity of β-cells, leading to an inability to compensate for the high demand for insulin in insulin-resistant states [17]. However, the role of senescence in relation to pancreatic β-cells is poorly understood, and there are no methods targeting the age-related aspect of the disease. β-Cells can compensate for increased metabolic demands by increasing insulin secretion, thus restraining hyperglycemia. This compensation may be limited by the age-related decline in β-cell proliferation, as shown in rodents [18] and humans [19]. This deficit in the proliferative response to increased demand may in part arise from the accumulation of senescent β-cells. Thus, with age, the accumulation of dysfunctional senescent β-cells likely contributes to impaired glucose tolerance and diabetes mellitus. However, the particular roles of β-cell senescence in the development of type 2 diabetes and the importance of β-cell senolysis as a potential therapy for suppressing T2D progression require more detailed discussion.
Chronic Inflammation as a Forerunner of Type 2 Diabetes
Inflammation primarily serves to restore homeostasis when the body is invaded by foreign pathogens or after trauma; if inflammation is limited, it plays a protective role in adaptation. In contrast, chronic inflammation may be a pathological condition that contributes to the development of various concomitant diseases. Indeed, chronic low-grade inflammation caused by metabolic stress due to constant nutrient overload leads to obesity, atherosclerosis, and T2D [20-22]. Many published studies have indicated immune involvement in the regulation of the consequences of metabolic inflammation. For example, macrophages play a role in pancreatic development [23, 24]. T2D is a chronic progressive disease associated with obesity and insulin resistance. The onset of T2D is primarily determined by the progressive inability of pancreatic β-cells to secrete enough insulin to maintain normoglycemia [25]. Many preclinical and clinical studies have shown a causal relationship between sterile low-grade inflammation and metabolic diseases, including T2D [20, 26-28]. Acute inflammation in response to pathogens and irritants usually begins at the site of injury and involves leukocytes, followed by a cellular phase dominated by granulocytes. In contrast, chronic inflammation in metabolic diseases and obesity does not involve an acute immunovascular response and mainly recruits mononuclear cells. In this case, a significant increase in proinflammatory cytokines and chemokines is usually observed in all tissues and organs, including pancreatic islets. In islets, the activation of the innate immune system may contribute to a decrease in β-cell mass and function [29, 30]. This activation is characterized by an increase in innate immune responses and the accumulation of inflammatory mediators. Before the first reports showing toxic effects of glucose on human pancreatic islets [31], a study using the diabetes-prone gerbil Psammomys obesus showed that hyperglycemia was associated with islet destruction in vivo [32]. Subsequently, glucose, fatty acids, and Toll-like receptor (TLR) agonists were shown to induce the expression of chemokines and proinflammatory cytokines in mouse islet cells [33-38]. The main function of β-cells is to produce and secrete insulin in response to metabolic demands. Importantly, β-cells have a highly developed endoplasmic reticulum (ER), where proinsulin is properly folded [39]. However, the prolonged high demand observed in the prediabetic state can lead to ER stress and subsequent inflammation [40]. In addition, local resident macrophages have been shown to respond to β-cell activity (actually, to ATP cosecreted with insulin by β-cells), leading to macrophage activation and an inflammatory response [41]. In addition, IL-1 receptor signaling has been shown to be an important part of metabolic syndrome [42, 43]. Thus, it has now been established that insulitis represents a partial decrease in the number and activity of mononuclear cells and an increase in the level of proinflammatory factors, leading to the development of T2D (Fig. 1).
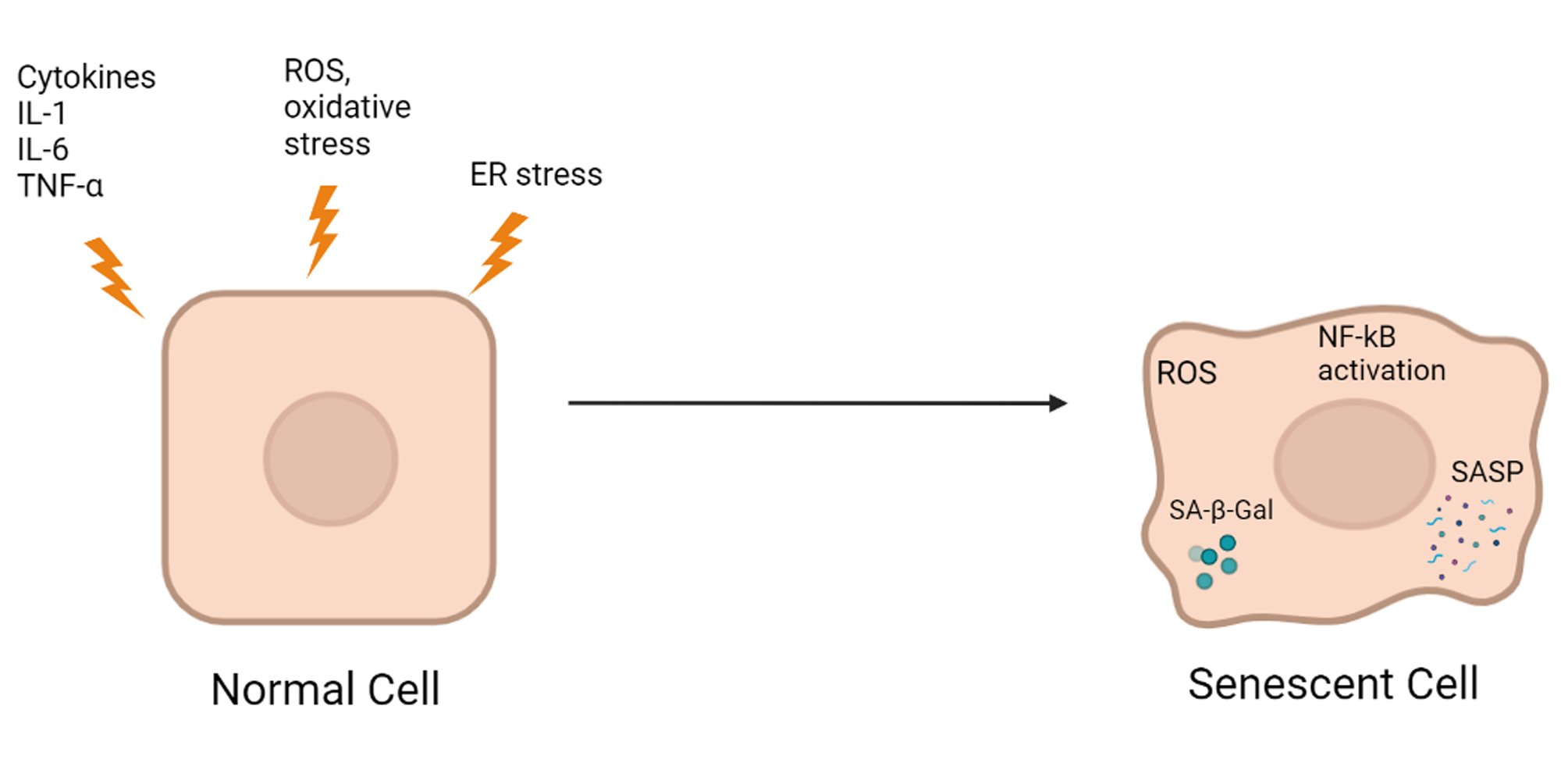
Fig. 1: The effects of inflammation, oxidative stress, and ER stress on β-cell senescence.
In addition, in studies of the IL-1 system in humans and rodents, the IL-1 system was shown to induce islet inflammation [31], which could be reduced using an IL-1 antagonist [44, 45]. However, many questions still need to be addressed in future studies, particularly to what extent anti-inflammatory treatments for T2D can be clinically implemented.
Β-Cell Aging and Type 2 Diabetes
Cell senescence is a condition in which cells stop dividing but remain metabolically active with an altered phenotype [46]. There are no universal markers of aging, and known markers do not match in different aging tissues. Senescent cells are related to many age-related diseases [47, 48]. There are several mechanisms underlying both natural and accelerated aging, both of which can manifest in age-related diseases; as such, “aging” and “age-related disease” can be seen as indistinguishable processes [49]. “Natural aging” usually refers to the gradual decline in biological functions throughout the whole life of an organism, while accelerated aging is likely associated with the same processes but runs at a faster rate due to additional stresses. For example, smoking, exposure to toxins, chemotherapy, a high-fat diet, and infectious diseases can all cause accelerated cell aging. Accelerated aging often affects particular tissues and organs rather than the body as a whole. For example, chronic obstructive pulmonary disease (COPD) caused by smoking is likely a result of accelerated lung aging caused by cigarette smoke [50]. The senescence of cells can potentially lead to biological dysfunction due to their inability to proliferate, the secretion of proinflammatory factors, a negative impact on neighboring cells, and protease-mediated degradation of extracellular components [15, 51]. Cell turnover is vital for replacing damaged cells, and it occurs throughout the life of an organism. Thus, permanently stopping senescent cells reduces the capability of tissue to regenerate. Such a decline in the regenerative capacity will affect both the growth of competent somatic tissues and post-mitotic tissues that depend on these growth-competent cells to maintain their normal state. Since pancreatic β-cells play an essential role in insulin production [52], it is not surprising that the dysfunction of these cells makes a major contribution to the pathogenesis of type 2 diabetes. Pancreatic islet cell dysfunction leading to impaired insulin secretion may be associated with a range of factors, including chronic inflammation, autoimmunity, oxidative stress, and ER stress [53]. It is known that inflammatory factors, such as cytokines, can also increase the senescence of neighboring cells [54]. Importantly, senescent cells are proinflammatory, lead to an immune response, demonstrate an increased level of ROS, and in some cases are capable of causing ER stress [47]. One of the pioneering studies indicating that pancreatic β-cell senescence may play a role as an underlying cause of type 2 diabetes was conducted using a high-fat diet in mice [55]. In this study, after 12 months of a high-fat diet, the proportion of β-cells determined by Ki67 staining was reduced by one-third compared to that of controls, while the proportion of cells positively stained with SA-beta-Gal increased strongly, indicating an increase in the proportion of senescent β-cells. The partial reversibility of β-cell senescence [56] suggests that this is a nonbinary phenomenon. External adverse factors may stimulate the induction of a subpopulation of senescent β-cells, while the gradual increase in the number of damaged β-cells likely accelerates the development of T2D. Ultimately, the accumulation of senescent β-cells may exceed the threshold, causing long-term metabolic dysfunction due to permanent loss of β-cell mass and function. Deletion of senescent β-cells or the reversal of senescence in a whole subpopulation of β-cells may interrupt this cascade of dysfunctions [57]. To use these therapeutic strategies, it is necessary to characterize the various subpopulations of senescent β-cells and the temporal patterns of the expression of senescence-associated genes. These experiments will also elucidate how senolytic treatments affect β-cell mass. Considering the central role of β-cell mass reduction in T2D, the restoration of the proliferative capacity of the cell population may provide a compensatory increase in insulin production to reduce blood glucose levels [58]. Senolysis induces a short-term decrease in the total number of β-cells, actively removing the senescent population. However, the effect of treatment on β-cell function and glucose tolerance indicates that, in the long term, senolysis may restore the proliferative capacity of the remaining β-cells by avoiding glucose toxicity or by reducing the local senescence-associated phenotype (SASP) [59]. These SASP factors may explain why the accumulation of senescent β-cells impairs glucose tolerance and β-cell function. It is necessary to evaluate the secretory phenotypes of various subpopulations of senescent β-cells to determine the specific mechanisms underlying the senescence-associated loss of mass and function of β-cells. Moreover, the characterization of the SASP of β-cells and the effects of its constituent factors may reveal additional therapeutic targets for T2D [60]. Insulin-like growth factor 1 receptor (IGF1R) has previously been identified as a novel marker for senescent β-cells [57]. Accounting for a high prevalence of diabetes in elderly individuals, impaired adaptive β-cell proliferation may likely be involved in the pathogenesis of type 2 diabetes mellitus in this population. This hypothesis is based on the fact that aging is often accompanied by insulin resistance [48]. In this regard, revealing a cause of reduced adaptive β-cell proliferation in aged animals is critical to alleviate the pathophysiological consequences of type 2 diabetes that affect elderly individuals. However, the roles of intrinsic factors, including senescent β-cells, and extrinsic factors of the aging systemic environment, such as signals from other organs, in impaired adaptive β-cell proliferation remain to be clarified. Signals from the liver to pancreatic β-cells are mediated by neural pathways [61, 62]. Different tissues and cell types are known to have different mechanisms of aging, described as hallmarks of aging [63]. Among the nine described features of cellular aging, SASP is one of the main characteristics of β-cell senescence [15]. Cell senescence is a stress response to a range of factors, such as DNA damage, ER stress, or oncogene activation. The senescent state is characterized by the arrest of cell proliferation, increased β-galactosidase (β-Gal) activity and SASP secretion. SASP proteins include soluble and insoluble factors, such as chemokines, cytokines, and extracellular matrix remodeling factors. These factors may induce dysfunction of surrounding cells and accelerate their senescence [64]; by recruiting immune cells, they contribute to the formation of a proinflammatory microenvironment. In addition, antiapoptotic pathways are activated in senescent cells, making them resistant to apoptosis. Senescence, as a response to stress, can occur at any time. However, the number of cellular stressors increases with age, while the immune response decreases, which may lead to the accumulation of senescent cells in the tissues of old animals. Senescent β-cells accumulate in the islets of aged mice and humans, and their proportion is further increased in conditions of insulin resistance and type 2 diabetes mellitus [63]. Although not an indicator of function per se, changes in β-cell mass due to a shift in the balance between proliferation and apoptosis affect β-cell functionality. In humans, the mass of β-cells normally does not change with aging [65]. Furthermore, some studies have shown that there is a functional reserve of β-cell mass that maintains the overall level of insulin secretion and blood glucose levels if the threshold is not reached. This functional reserve is estimated as 20–25% in rats [66] and 50–70% in humans based on data from newly diagnosed type 1 [67] or type 2 [68] diabetes subjects. However, aging reduces the ability of β-cells to proliferate in response to higher metabolic demands in rodents [69] and humans [19], leading to a limitation in the regenerative capacity of β-cells. In old mice, β-cell regeneration is suppressed for a long time [18], which may be associated with β-cell senescence [70]. This notion was confirmed by the knockout of p16Ink4a (a marker and effector of senescence), which caused increased β-cell proliferation in aged mice [71]. However, the mass of β-cells in rodents increases with age [72], which means that the actual pool of replicating β-cells is larger in adults than in young animals [73]. Generally, we may conclude that age-related changes in β-cell mass and proliferation do not affect β-cell performance under nondiabetic conditions. Peripheral insulin sensitivity plays a key role in β-cell functioning and the development of type 2 diabetes. The condition of insulin resistance caused by obesity or a sedentary lifestyle is initially accompanied by a compensatory increase in insulin secretion, which may subsequently decline and translate into overt diabetes in susceptible individuals [17]. It is generally accepted that insulin resistance occurs with age [74], in part due to the accumulation of senescent cells in adipose tissue and subsequent local secretion of SASP. Interestingly, insulin sensitivity has been found to improve with age [75], even though insulin secretion steadily declines. This finding indicates the heterogeneity of aging across individuals, which is a key concept of the pathophysiology of diabetes, describes as five subgroups of adult diabetes that differ in the degree of β-cell dysfunction versus insulin resistance [76]. The clinically defined subgroup to which an individual belongs may be used for personalized treatment (Fig. 2).
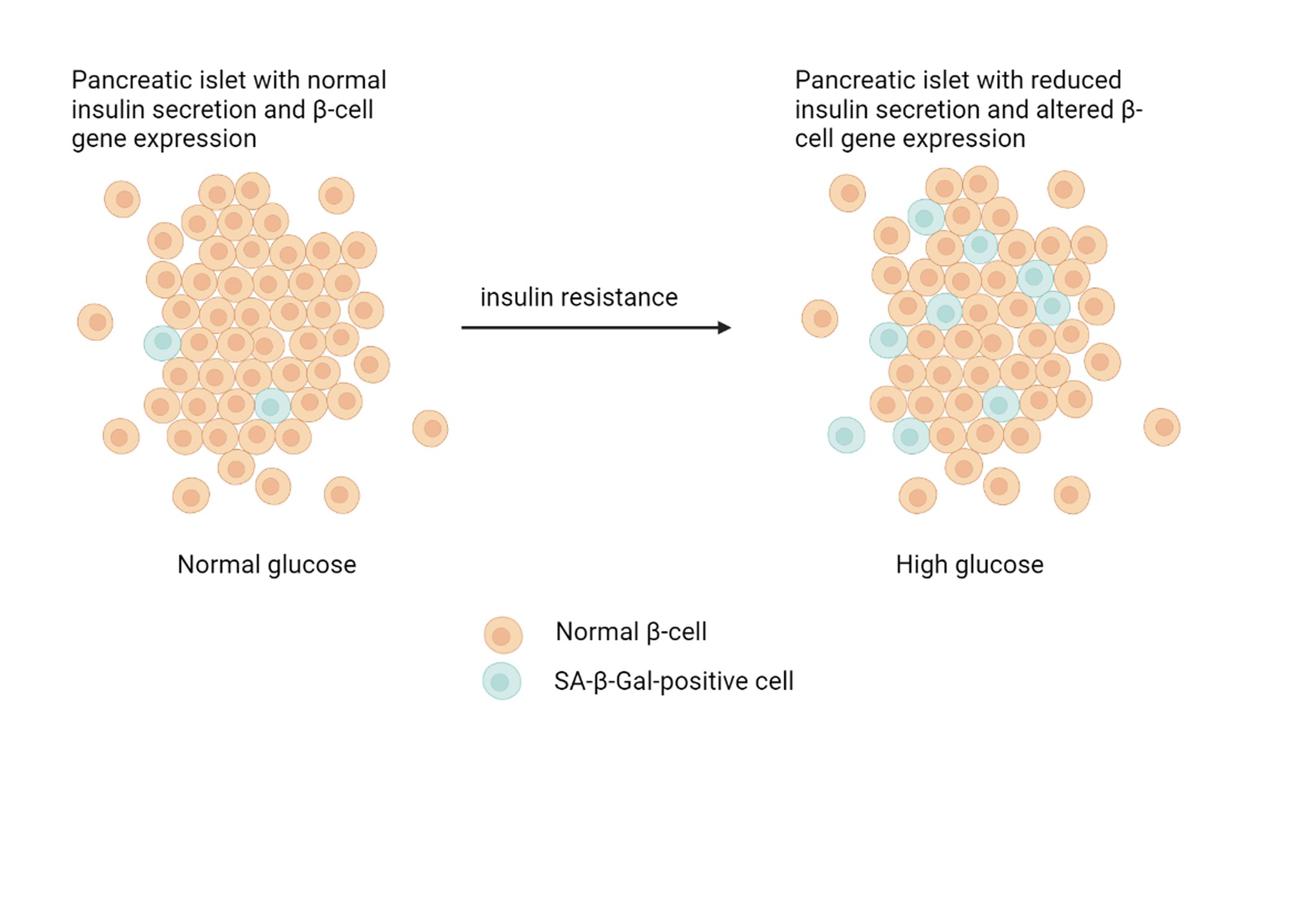
Fig. 2: Insulin resistance-accelerated β-cell senescence leading to loss of function and cellular identity.
Changes in other cell types, such as blood vessels and exocrine cells, may also affect insulin secretion. For example, revascularization of pancreatic islets of aged mice using vessels from young mice has been shown to restore their functional and proliferative capacities [77]. In addition, circulating factors provide other interesting insights into the mechanisms by which insulin secretion is regulated. Thus, studies using parabiosis have shown that the replicative capacity of pancreatic islets of old mice is restored when parabiosis is performed with young mice [58], suggesting the existence of a circulating factor(s) that “rejuvenates” β-cells.
Therapeutic Approaches to Reduce Cell Senescence
Cellular senescence involves fundamental changes in gene expression and proliferative arrest. Senescence may be caused by stresses, such as DNA damage, telomere shortening, oncogenic mutations, metabolic and mitochondrial dysfunction, inflammation, and autoimmune conditions [46, 78, 79]. The mass of senescent cells increases in many tissues with age, in pathological loci during various chronic diseases, and after irradiation or chemotherapy [80]. Senescent cells secrete proinflammatory cytokines, chemokines, proteases, and other factors called SASP [81, 82]. Senescent cells likely play a role in the development and progression of metabolic diseases, such as obesity and type 2 diabetes, which in turn can lead to diabetes-induced cell senescence and the development of other diabetes-associated diseases, such as cardiovascular and renal diseases. As a result of the changes in gene expression, cell function and proliferation are impaired, which also affect (through SASP) intercellular signaling [64]. Therefore, targeting senescent cells has great potential to improve health [49, 83]. Such strategies may include the prevention of cell senescence, reversion of senescence, inhibition of some aspects of the senescent phenotype, and elimination of senescent cells (senolysis). Regardless, reversing the signs of cellular senescence is a potential new approach to the treatment of T2D. The senolytic drug ABT263 (a specific inhibitor of the antiapoptotic proteins BCL-2 and BCL-xL) was shown to selectively kill senescent cells in culture in a cell-type-independent manner by inducing apoptosis. Oral administration of ABT263 to either young adult mice or mice exposed to a sublethal dose of radiation effectively reduced the number of senescent cells, including senescent bone marrow hematopoietic stem cells and senescent muscle stem cells [84]. A new panel of small molecules has been developed based on resveratrol, which was previously proposed for targeting mRNA splicing, to determine whether altered splicing factor expression could affect replicative senescence features [85]. The authors showed that under growth-permissive conditions, senescent cells showed restored splicing factor expression, which led to an increase in telomere length, re-entry into the cell cycle, and restoration of proliferation. This study was the first demonstration that restoring the levels of splicing factors using resveratrol analogs (resveralogs) was associated with the inhibition of cellular senescence in primary human fibroblasts. Thus, low-molecular-weight modulators of such targets may be promising new anti-degenerative therapies. Another therapeutic approach is the inhibition of damaging components of the senile phenotype, primarily, the pro-inflammatory secretome. In recent years, regulation of the aging secretome has been actively studied, which has made it possible to inhibit it with drugs [60]. As an example of the potential benefits, one study demonstrated that inhibition of the senescent secretome reduced senile frailty [82]. Eliminating senescent cells in mice by inducing cell death has provided the most compelling evidence that senescent cells play a role in diseases and that their removal may be beneficial to health [86]. Thus, in recent years, several substances have been identified or created that can specifically kill senescent cells [87-89]. In addition to the use of pharmacological substances, the immune system can be used to remove senescent cells in a similar way. For example, immunotherapy is currently being used to fight some types of cancer [90]. In addition, in some cases, a beneficial effect of humoral factors [91] that regulate the adaptive proliferation of B cells was shown. Recently, senolytic therapies specifically targeting senescent cells were shown to be beneficial for a variety of age-related conditions, such as hepatic steatosis, stem cell biology, and longevity [59, 84]. T2D usually develops in response to overeating and lack of physical activity in patients with a predisposition to insulin resistance and β-cell dysfunction. If compensation for insulin resistance does not occur, β-cell dysfunction develops [17, 52]. As shown in rodents, mechanisms to compensate for increased insulin resistance include increased secretion of insulin [17] and β-cell proliferation [92]. The chronological age of animals limits the proliferative capacity of β-cells [51, 93], most likely through mechanisms associated with cell senescence. Treatment for one month with JAK1/2 inhibitors or rapamycin was shown to enhance cell functions in aged mice [82, 94], and JAK1/2 inhibitors and rapamycin have a broad spectrum of action, including the suppression of some SASP constituents. These data indicate that senescent cells can potentially contribute to age-related physical dysfunction and that appropriate therapies can increase lifespan and health (Fig. 3).
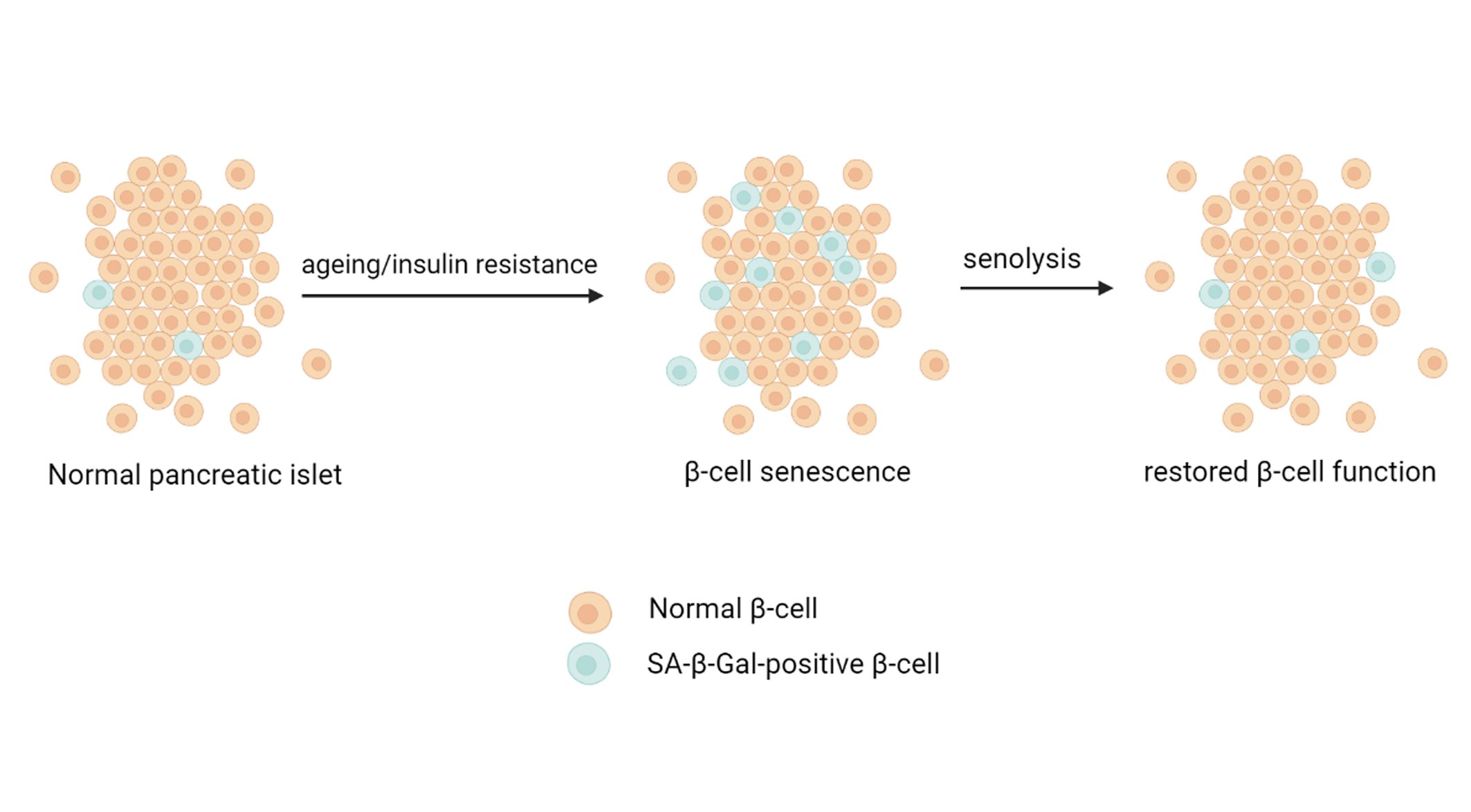
Fig. 3: Aging and insulin resistance accelerate β-cell senescence through increasing SASP, whereas β-cell senolytic therapies restore β-cell function.
Transcriptome analysis revealed increased expression of prosurvival networks in senescent cells, consistent with their established resistance to apoptosis. The use of siRNA to silence the expression of key nodes of this network, including ephrins (EFNB1 or 3), PI3Kδ, p21, BCL-xL, or plasminogen-activated inhibitor-2, killed senescent cells but not proliferating or quiescent, differentiated cells. Drugs targeting these factors selectively kill senescent cells. Dasatinib eliminated senescent human fat progenitor cells, while quercetin was more effective against senescent human endothelial cells [95]. Finally, there is strong evidence for oxidative stress in both type 1 and type 2 diabetes [10-12]. In addition, the increased production of reactive oxygen or nitrogen species led to the increased consumption of endogenous antioxidant enzymes (superoxide dismutase, glutathione peroxidase and catalase) and decreased levels of some low-molecular-weight antioxidants, such as vitamin D [13]. Notably, pancreatic β-cells have the greatest constitutive vulnerability to reactive oxygen species (ROS). Indeed, compared to levels of other mammalian tissues, pancreatic β-cells contain lower levels of antioxidant enzymes, such as superoxide dismutase, catalase, and glutathione peroxidase, making these cells more vulnerable to the damaging effects of ROS [96, 97]. More than three decades ago, it was shown that glutathione peroxidase activity and resistance to peroxide are approximately 20 times higher in the liver and kidney than in the pancreas [98]. Naturally, in conditions of reduced activity of endogenous antioxidant enzymes, more attention is given to new proteins, such as peroxiredoxin 6 (Prdx6), which also have antioxidant activity and may be able to protect pancreatic β-cells during the development of diabetes. We have conducted systemic studies of the role of Prdx6 in protection against type 1 diabetes in vivo and in vitro. Interestingly, the insulin-stimulating activity of Prx6 in vitro was detected both when RIN-m5F β-cells were cultivated under normal conditions and under stressful conditions, inducing cell death [99]. In an alloxan- or streptozotocin-induced mouse model of diabetes, intravenous administration of recombinant Prdx6 prevented hyperglycemia, reduced mortality, restored the plasma cytokine profile, and reduced β-cell destruction in the islets of Langerhans in the mouse pancreas [100-102]. Interestingly, a group of authors from Italy, using PRDX6 knockout mice, proved that Prdx6 is involved in the pathogenesis of type 2 diabetes, which was characterized by both reduced glucose-dependent insulin secretion and increased insulin resistance [103-104]. We suggest that these results are attributable to the ability of Prdx6 to stimulate insulin production in pancreatic β-cells [99], which should clearly affect the insulin resistance characteristic of type 2 diabetes. In addition, using 3T3 fibroblasts, Prdx6 reduced cell senescence caused by a sublethal dose of X-ray radiation [105]. This finding indicates that Prdx6 may be included in the list of senolytics. In conclusion, preclinical studies are needed to examine the potential toxicity of senolytics and to develop regimens for their administration. Revealing the possible side effects of senolytics as a class and as individual substances is of particular importance.
Conclusions and Directions
Due to the increase in life expectancy and affluence, aging-associated metabolic disorders such as obesity and type 2 diabetes increase rapidly and become a serious health burden. In 2021, the estimated prevalence of patients with diabetes mellitus reached 537 million, and diabetes mellitus was responsible for 6.7 million deaths [1]. Increased insulin resistance and impaired insulin secretion are significant characteristics manifested by patients with type 2 diabetes mellitus, and this is associated with accumulation of senescent cells in multiple organs. Cellular senescence is defined as a state of irreversible cell cycle arrest with concomitant functional decline [106].
Targeting senescent cells offers a lot of therapeutic opportunities, particularly in the context of age-associated diseases. This knowledge has fueled testing of senolytic drugs as a novel therapeutic paradigm. Increased insulin resistance and impaired insulin secretion have central roles in the pathophysiology of T2DM [107]. Age-associated changes in β cell functions and proliferation suggest the importance of senescence-related aspects of pancreatic β cells in pathogenesis of T2DM.
Senescence is a cellular state characterized by irreversible cell cycle arrest with functional decline due to telomere shortening or senescence-inducing stresses, e.g., DNA damage, oncogenic stress, and oxidative stress. As research targeting “cellular senescence/chronic inflammation” is developing rapidly, removal of senescent cells, called “senolysis”, is a potentially attractive approach [108].
Although many of the detrimental consequences of senescence may be attributed to SASP, direct evidences of the SASP role are often lacking. Transgenic models with specific targeting of senescent cells may prove the causality.
Senescent cells undergo a permanent growth arrest, produce a complex secretome (known as the senescence-associated secretory phenotype, SASP), and develop characteristic changes including transcriptional, epigenetic, morphological, and metabolic alterations [109]. The development of ingenious models allowing the manipulation of the SASP will help to determine its precise involvement as a mediator of the effects exerted by senescent cells. Actually, it may be very complicated given SASP heterogeneity and its context-dependent effects.
It is also important to reveal regulators of the SASP and discriminate the SASP from inflammatory events induced by other factors. For this purpose, identification of reliable in vivo markers of senescence and models that allow for targeted manipulation of SASP regulatory factors within senescent cells may be useful. Indeed, a key function of the SASP is signaling for different immune cells, including natural killer (NK) cells, macrophages, and T cells. Immune-mediated clearance of senescent cells also suppresses tumor initiation [110].
To design senotherapies targeted to specific diseases, we need better understanding as to what cell types undergo senescence, because eliminating certain senescent cell types can be harmful [111, 112]. Clarification of the molecular and physiological properties of senescent cells, including their SASP profile, is also needed.
There is every reason to believe that beta-cells senescence and associated consequences may have some genetic basis. Indeed, diabetes mellitus was reported to co-exist with progeria [113]. Progeria (premature aging) or Werner syndrome (WS), also known as adult progeroid syndrome, is a rare, early-onset, and age-related disease characterized by segments of aging phenotypes [113]. In addition, it was shown that partial lipodystrophy with severe insulin resistance may manifest in WS-linked premature aging syndrome [114].
Anyway, targeting senescent cells to delay aging and limit dysfunction, known as "senotherapy," is gaining momentum, and drugs that selectively kill senescent cells, termed "senolytics" are a major focus. A little explored area related to the clinical use of senolytics needs to be advanced.
Further investigations of molecular mechanisms of cellular senescence in diabetes and senolytic compounds with clinical efficacy and safety should provide a clinical application of this concept.
Acknowledgements
This work was supported by the Russian Scientific Foundation, project 23-24-00041. The authors thank AJE for help with language editing.
Disclosure Statement
The authors have no conflicts of interest to declare
References
| 1 | King H, Aubert RE, Herman WH: Global burden of diabetes, 1995-2025: Prevalence, numerical estimates, and projections. Diabetes Care 1998;21:1414-1431. https://doi.org/10.2337/diacare.21.9.1414 |
| 2 | IDF Diabetes Atlas. 9th ed. Brussels: International Diabetes Federation, 2019. |
| 3 | Kleinert M, Clemmensen C, Hofmann SM, Moore MC, Renner S, Woods SC, Huypens P, Beckers J, de Angelis MH, Schürmann A, Bakhti M, Klingenspor M, Heiman M, Cherrington AD, Ristow M, Lickert H, Wolf E, Havel PJ, Müller TD, Tschöp MH: Animal models of obesity and diabetes mellitus. Nat Rev Endocrinol 2018;14:140-162. https://doi.org/10.1038/nrendo.2017.161 |
| 4 | Zhuo J, Zeng Q, Cai D, Zeng X, Chen Y, Gan H, Huang X, Yao N, Huang D, Zhang C: Evaluation of type 2 diabetic mellitus animal models via interactions between insulin and mitogen activated protein kinase signaling pathways induced by a high fat and sugar diet and streptozotocin. Mol Med Rep 2018;17:5132-5142. https://doi.org/10.3892/mmr.2018.8504 |
| 5 | Pearson ER: Type 2 diabetes: a multifaceted disease. Diabetologia 2019;62:1107-1112. https://doi.org/10.1007/s00125-019-4909-y |
| 6 | Dadheech N, Srivastava A, Paranjape N, Gupta S, Dave A, Shah GM, Bhonde RR, Gupta S: Swertisin an anti-diabetic compound facilitate islet neogenesis from pancreatic stem/progenitor cells via p-38 MAP kinase-SMAD pathway: an in-vitro and in-vivo study. PLoS One 2015;10:e0128244. https://doi.org/10.1371/journal.pone.0128244 |
| 7 | Christensen AA, Gannon M: The beta cell in type 2 diabetes. Curr Diab Rep 2019;19:81. https://doi.org/10.1007/s11892-019-1196-4 |
| 8 | Rutter GA, Pullen TJ, Hodson DJ, Martinez-Sanchez A: Pancreatic β-cell identity, glucose sensing and the control of insulin secretion. Biochem J 2015;466:203-218. https://doi.org/10.1042/BJ20141384 |
| 9 | Miranda-Díaz AG, Pazarín-Villaseñor L, Yanowsky-Escatell FG, Andrade-Sierra J: Oxidative stress in diabetic nephropathy with early chronic kidney disease. J Diabetes Res 2016;2016:7047238. https://doi.org/10.1155/2016/7047238 |
| 10 | Maiese K: New insights for oxidative stress and diabetes mellitus. Oxid Med Cell Longev 2015;2015:875961. https://doi.org/10.1155/2015/875961 |
| 11 | Savastio S, Cadario F, Genoni G, Bellomo G, Bagnati M, Secco G, Picchi R, Giglione E, Bona G: Vitamin D deficiency and glycemic status in children and adolescents with type 1 diabetes mellitus. PLoS One 2016;11:e0162554. https://doi.org/10.1371/journal.pone.0162554 |
| 12 | Ceriello A: New insights on oxidative stress and diabetic complications may lead to a "causal" antioxidant therapy. Diabetes Care 2003;26:1589-1596. https://doi.org/10.2337/diacare.26.5.1589 |
| 13 | Boudina S, Abel ED: Diabetic cardiomyopathy revisited. Circulation 2007;115:3213-3223. https://doi.org/10.1161/CIRCULATIONAHA.106.679597 |
| 14 | Lankin VZ, Sharapov MG, Goncharov RG, Tikhaze AK, Novoselov VI: Natural dicarbonyls inhibit peroxidase activity of peroxiredoxins. Dokl Biochem Biophys 2019;485:132-134. https://doi.org/10.1134/S1607672919020157 |
| 15 | Aguayo-Mazzucato C, Andle J, Lee TB Jr, Midha A, Talemal L, Chipashvili V, Hollister-Lock J, van Deursen J, Weir G, Bonner-Weir S: Acceleration of β cell aging determines diabetes and senolysis improves disease outcomes. Cell Metab 2019;30:129-142. https://doi.org/10.1016/j.cmet.2019.05.006 |
| 16 | Kirkland JL, Tchkonia T: Senolytic drugs: from discovery to translation. J Intern Med 2020;288:518-536. https://doi.org/10.1111/joim.13141 |
| 17 | Weir GC, Bonner-Weir S: Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 2004;53:16-21. https://doi.org/10.2337/diabetes.53.suppl_3.S16 |
| 18 | Stolovich-Rain M, Hija A, Grimsby J, Glaser B, Dor Y: Pancreatic beta cells in very old mice retain capacity for compensatory proliferation. J Biol Chem 2012;287:27407-27414. https://doi.org/10.1074/jbc.M112.350736 |
| 19 | Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, Wright AJ, Atkinson MA, Rhodes CJ: Formation of a human β-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab 2012;97:3197-3206. https://doi.org/10.1210/jc.2012-1206 |
| 20 | Donath MY, Shoelson SE: Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 2011;11:98-107. https://doi.org/10.1038/nri2925 |
| 21 | Wellen KE, Hotamisligil GS: Inflammation, stress, and diabetes. J Clin Invest 2005;115:1111-1119. https://doi.org/10.1172/JCI25102 |
| 22 | Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H: Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003;112:1821-1830. https://doi.org/10.1172/JCI200319451 |
| 23 | Geutskens SB, Otonkoski T, Pulkkinen MA, Drexhage HA, Leenen PJ: Macrophages in the murine pancreas and their involvement in fetal endocrine development in vitro. J Leukoc Biol 2005;78:845-852. https://doi.org/10.1189/jlb.1004624 |
| 24 | Banaei-Bouchareb L, Peuchmaur M, Czernichow P, Polak M: A transient microenvironment loaded mainly with macrophages in the early developing human pancreas. J Endocrinol 2006;188:467-480. https://doi.org/10.1677/joe.1.06225 |
| 25 | Donath MY, Böni-Schnetzler M, Ellingsgaard H, Ehses JA: Islet inflammation impairs the pancreatic beta-cell in type 2 diabetes. Physiology (Bethesda) 2009;24:3253-3231. https://doi.org/10.1152/physiol.00032.2009 |
| 26 | Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY: Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 2007;356:1517-1526. https://doi.org/10.1056/NEJMoa065213 |
| 27 | Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ; CANTOS Trial Group: Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119-1131. https://doi.org/10.1056/NEJMoa1707914 |
| 28 | De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD, Le O, Beauséjour C, Ambati J, Ambati K, Simon M, Seluanov A, Gorbunova V, Slagboom PE, Helfand SL, Neretti N, Sedivy JM; L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019;566:73-78. https://doi.org/10.1038/s41586-018-0784-9 |
| 29 | Donath MY, Halban PA: Decreased beta-cell mass in diabetes: significance, mechanisms and therapeutic implications. Diabetologia 2004;47:581-589. https://doi.org/10.1007/s00125-004-1336-4 |
| 30 | Donath MY, Dalmas É, Sauter NS, Böni-Schnetzler M: Inflammation in obesity and diabetes: islet dysfunction and therapeutic opportunity. Cell Metab 2013;17:860-872. https://doi.org/10.1016/j.cmet.2013.05.001 |
| 31 | Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, Donath MY: Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 2002;110:851-860. https://doi.org/10.1172/JCI200215318 |
| 32 | Donath MY, Gross DJ, Cerasi E, Kaiser N: Hyperglycemia-induced beta-cell apoptosis in pancreatic islets of Psammomys obesus during development of diabetes. Diabetes 1999;48:738-744. https://doi.org/10.2337/diabetes.48.4.738 |
| 33 | Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, Gueripel X, Ellingsgaard H, Schneider MK, Biollaz G, Fontana A, Reinecke M, Homo-Delarche F, Donath MY: Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 2007;56:2356-2370. https://doi.org/10.2337/db06-1650 |
| 34 | Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, Yagi N, Ohto U, Kimoto M, Miyake K, Tobe K, Arai H, Kadowaki T, Nagai R: Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab 2012;15:518-533. https://doi.org/10.1016/j.cmet.2012.01.023 |
| 35 | Böni-Schnetzler M, Boller S, Debray S, Bouzakri K, Meier DT, Prazak R, Kerr-Conte J, Pattou F, Ehses JA, Schuit FC, Donath MY: Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology 2009;150:5218-5229. https://doi.org/10.1210/en.2009-0543 |
| 36 | Ehses JA, Meier DT, Wueest S, Rytka J, Boller S, Wielinga PY, Schraenen A, Lemaire K, Debray S, Van Lommel L, Pospisilik JA, Tschopp O, Schultze SM, Malipiero U, Esterbauer H, Ellingsgaard H, Rütti S, Schuit FC, Lutz TA, Böni-Schnetzler M, Konrad D, Donath MY: Toll-like receptor 2-deficient mice are protected from insulin resistance and beta cell dysfunction induced by a high-fat diet. Diabetologia 2010;53:1795-1806. https://doi.org/10.1007/s00125-010-1747-3 |
| 37 | Nackiewicz D, Dan M, He W, Kim R, Salmi A, Rütti S, Westwell-Roper C, Cunningham A, Speck M, Schuster-Klein C, Guardiola B, Maedler K, Ehses JA: TLR2/6 and TLR4-activated macrophages contribute to islet inflammation and impair beta cell insulin gene expression via IL-1 and IL-6. Diabetologia 2014;57:1645-1654. https://doi.org/10.1007/s00125-014-3249-1 |
| 38 | Westwell-Roper C, Denroche HC, Ehses JA, Verchere CB: Differential activation of innate immune pathways by distinct islet amyloid polypeptide (IAPP) aggregates. J Biol Chem 2016;291:8908-8917. https://doi.org/10.1074/jbc.M115.712455 |
| 39 | Dodson G, Steiner D: The role of assembly in insulin's biosynthesis. Curr Opin Struct Biol 1998;8:189-194. https://doi.org/10.1016/S0959-440X(98)80037-7 |
| 40 | Oslowski CM, Hara T, O'Sullivan-Murphy B, Kanekura K, Lu S, Hara M, Ishigaki S, Zhu LJ, Hayashi E, Hui ST, Greiner D, Kaufman RJ, Bortell R, Urano F: Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab 2012;16:265-273. https://doi.org/10.1016/j.cmet.2012.07.005 |
| 41 | Weitz JR, Makhmutova M, Almaça J, Stertmann J, Aamodt K, Brissova M, Speier S, Rodriguez-Diaz R, Caicedo A: Mouse pancreatic islet macrophages use locally released ATP to monitor beta cell activity. Diabetologia 2018;61:182-192. https://doi.org/10.1007/s00125-017-4416-y |
| 42 | Westwell-Roper CY, Chehroudi CA, Denroche HC, Courtade JA, Ehses JA, Verchere CB: IL-1 mediates amyloid-associated islet dysfunction and inflammation in human islet amyloid polypeptide transgenic mice. Diabetologia 2015;58:575-585. https://doi.org/10.1007/s00125-014-3447-x |
| 43 | Westwell-Roper CY, Ehses JA, Verchere CB: Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1β production and β-cell dysfunction. Diabetes 2014;63:1698-1711. https://doi.org/10.2337/db13-0863 |
| 44 | Ehses JA, Lacraz G, Giroix MH, Schmidlin F, Coulaud J, Kassis N, Irminger JC, Kergoat M, Portha B, Homo-Delarche F, Donath MY: IL-1 antagonism reduces hyperglycemia and tissue inflammation in the type 2 diabetic GK rat. Proc Natl Acad Sci U S A 2009;106:13998-14003. https://doi.org/10.1073/pnas.0810087106 |
| 45 | Sauter NS, Schulthess FT, Galasso R, Castellani LW, Maedler K: The antiinflammatory cytokine interleukin-1 receptor antagonist protects from high-fat diet-induced hyperglycemia. Endocrinology 2008;149:2208-2218. https://doi.org/10.1210/en.2007-1059 |
| 46 | Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL: Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 2013;123:966-972. https://doi.org/10.1172/JCI64098 |
| 47 | Burton DG, Faragher RG: Cellular senescence: from growth arrest to immunogenic conversion. Age (Dordr) 2015;37:27. https://doi.org/10.1007/s11357-015-9764-2 |
| 48 | Sikora E, Bielak-Zmijewska A, Mosieniak G: Cellular senescence in ageing, age-related disease and longevity. Curr Vasc Pharmacol 2014;12:698-706. https://doi.org/10.2174/1570161111666131219094045 |
| 49 | Faragher RG: Should we treat aging as a disease? The consequences and dangers of miscategorisation. Front Genet 2015;6:171. https://doi.org/10.3389/fgene.2015.00171 |
| 50 | Mercado N, Ito K, Barnes PJ: Accelerated ageing of the lung in COPD: new concepts. Thorax 2015;70:482-489. https://doi.org/10.1136/thoraxjnl-2014-206084 |
| 51 | Aguayo-Mazzucato C: Functional changes in beta cells during ageing and senescence. Diabetologia 2020;632022-2029. https://doi.org/10.1007/s00125-020-05185-6 |
| 52 | Leahy JL: Pathogenesis of type 2 diabetes mellitus. Arch Med Res 2005;36:197-209. https://doi.org/10.1016/j.arcmed.2005.01.003 |
| 53 | Cerf ME: Beta cell dysfunction and insulin resistance. Front Endocrinol (Lausanne) 2013;4:37. https://doi.org/10.3389/fendo.2013.00037 |
| 54 | Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, Longerich T, Sansom OJ, Benitah SA, Zender L, Gil J: A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013;15:978-990. https://doi.org/10.1038/ncb2784 |
| 55 | Sone H, Kagawa Y: Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 2005;48:58-67. https://doi.org/10.1007/s00125-004-1605-2 |
| 56 | Campisi J, d'Adda di Fagagna F: Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007;8:729-740. https://doi.org/10.1038/nrm2233 |
| 57 | Aguayo-Mazzucato C, van Haaren M, Mruk M, Lee TB Jr, Crawford C, Hollister-Lock J, Sullivan BA, Johnson JW, Ebrahimi A, Dreyfuss JM, Van Deursen J, Weir GC, Bonner-Weir S: β cell aging markers have heterogeneous distribution and are induced by insulin resistance. Cell Metab 2017;25:898-910. https://doi.org/10.1016/j.cmet.2017.03.015 |
| 58 | Salpeter SJ, Khalaileh A, Weinberg-Corem N, Ziv O, Glaser B, Dor Y: Systemic regulation of the age-related decline of pancreatic β-cell replication. Diabetes 2013;62:2843-2848. https://doi.org/10.2337/db13-0160 |
| 59 | Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, Onken JL, Johnson KO, Verzosa GC, Langhi LGP, Weigl M, Giorgadze N, LeBrasseur NK, Miller JD, Jurk D, Singh RJ, Allison DB, Ejima K, Hubbard GB, Ikeno Y, Cubro H, Garovic VD, Hou X, Weroha SJ, Robbins PD, Niedernhofer LJ, Khosla S, Tchkonia T, Kirkland JL: Senolytics improve physical function and increase lifespan in old age. Nat Med 2018;24:1246-1256. https://doi.org/10.1038/s41591-018-0092-9 |
| 60 | Malaquin N, Martinez A, Rodier F: Keeping the senescence secretome under control: Molecular reins on the senescence-associated secretory phenotype. Exp Gerontol 2016;82:39-49. https://doi.org/10.1016/j.exger.2016.05.010 |
| 61 | Imai J, Katagiri H, Yamada T, Ishigaki Y, Suzuki T, Kudo H, Uno K, Hasegawa Y, Gao J, Kaneko K, Ishihara H, Niijima A, Nakazato M, Asano T, Minokoshi Y, Oka Y: Regulation of pancreatic beta cell mass by neuronal signals from the liver. Science 2008;322:1250-1254. https://doi.org/10.1126/science.1163971 |
| 62 | Yamamoto J, Imai J, Izumi T, Takahashi H, Kawana Y, Takahashi K, Kodama S, Kaneko K, Gao J, Uno K, Sawada S, Asano T, Kalinichenko VV, Susaki EA, Kanzaki M, Ueda HR, Ishigaki Y, Yamada T, Katagiri H: Neuronal signals regulate obesity induced β-cell proliferation by FoxM1 dependent mechanism. Nat Commun 2017;8:1930. https://doi.org/10.1038/s41467-017-01869-7 |
| 63 | López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G: The hallmarks of aging. Cell 2013;153:1194-1217. https://doi.org/10.1016/j.cell.2013.05.039 |
| 64 | Coppé JP, Desprez PY, Krtolica A, Campisi J: The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010;5:99-118. https://doi.org/10.1146/annurev-pathol-121808-102144 |
| 65 | Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC: β-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care 2013;36:111-117. https://doi.org/10.2337/dc12-0421 |
| 66 | Olsson R, Carlsson PO: A low-oxygenated subpopulation of pancreatic islets constitutes a functional reserve of endocrine cells. Diabetes 2011;60:2068-2075. https://doi.org/10.2337/db09-0877 |
| 67 | Damond N, Engler S, Zanotelli VRT, Schapiro D, Wasserfall CH, Kusmartseva I, Nick HS, Thorel F, Herrera PL, Atkinson MA, Bodenmiller B: A map of human type 1 diabetes progression by imaging mass cytometry. Cell Metab 2019;29:755-768. https://doi.org/10.1016/j.cmet.2018.11.014 |
| 68 | Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC: Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003;52:102-110. https://doi.org/10.2337/diabetes.52.1.102 |
| 69 | Scaglia L, Cahill CJ, Finegood DT, Bonner-Weir S: Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 1997;138:1736-1741. https://doi.org/10.1210/endo.138.4.5069 |
| 70 | Tschen SI, Dhawan S, Gurlo T, Bhushan A: Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes 2009;58:1312-1320. https://doi.org/10.2337/db08-1651 |
| 71 | Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE: p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006;443:453-457. https://doi.org/10.1038/nature05092 |
| 72 | Montanya E, Nacher V, Biarnés M, Soler J: Linear correlation between beta-cell mass and body weight throughout the lifespan in Lewis rats: role of beta-cell hyperplasia and hypertrophy. Diabetes 2000;49:1341-1346. https://doi.org/10.2337/diabetes.49.8.1341 |
| 73 | Aguayo-Mazzucato C, Bonner-Weir S: Pancreatic β cell regeneration as a possible therapy for diabetes. Cell Metab 2018;27:57-67. https://doi.org/10.1016/j.cmet.2017.08.007 |
| 74 | Ehrhardt N, Cui J, Dagdeviren S, Saengnipanthkul S, Goodridge HS, Kim JK, Lantier L, Guo X, Chen YI, Raffel LJ, Buchanan TA, Hsueh WA, Rotter JI, Goodarzi MO, Péterfy M: Adiposity-independent effects of aging on insulin sensitivity and clearance in mice and humans. Obesity (Silver Spring) 2019;27:434-443. https://doi.org/10.1002/oby.22418 |
| 75 | Xiao J, Weng J, Ji L, Jia W, Lu J, Shan Z, Liu J, Tian H, Ji Q, Yang Z, Yang W; China National Diabetes and Metabolic Disorders Study Group: Worse pancreatic β-cell function and better insulin sensitivity in older Chinese without diabetes. J Gerontol A Biol Sci Med Sci 2014;69:463-470. https://doi.org/10.1093/gerona/glt104 |
| 76 | Ahlqvist E, Storm P, Käräjämäki A, Martinell M, Dorkhan M, Carlsson A, Vikman P, Prasad RB, Aly DM, Almgren P, Wessman Y, Shaat N, Spégel P, Mulder H, Lindholm E, Melander O, Hansson O, Malmqvist U, Lernmark Å, Lahti K, Forsén T, Tuomi T, Rosengren AH, Groop L: Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol 2018;6:361-369. https://doi.org/10.1016/S2213-8587(18)30051-2 |
| 77 | Almaça J, Molina J, Arrojo E Drigo R, Abdulreda MH, Jeon WB, Berggren PO, Caicedo A, Nam HG: Young capillary vessels rejuvenate aged pancreatic islets. Proc Natl Acad Sci U S A 2014;111:17612-17617. https://doi.org/10.1073/pnas.1414053111 |
| 78 | Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, Gerencser AA, Verdin E, Campisi J: Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 2016;23:303-314. https://doi.org/10.1016/j.cmet.2015.11.011 |
| 79 | Kim YK, Sussel L, Davidson HW: Inherent beta cell dysfunction contributes to autoimmune susceptibility. Biomolecules 2021;11:512. https://doi.org/10.3390/biom11040512 |
| 80 | Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T: DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009;8:311-323. https://doi.org/10.1111/j.1474-9726.2009.00481.x |
| 81 | Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J: Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008;6:2853-2868. https://doi.org/10.1371/journal.pbio.0060301 |
| 82 | Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, White TA, Johnson KO, Stout MB, Mezera V, Giorgadze N, Jensen MD, LeBrasseur NK, Kirkland JL: JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A 2015;112:6301-6310. https://doi.org/10.1073/pnas.1515386112 |
| 83 | Gurău F, Baldoni S, Prattichizzo F, Espinosa E, Amenta F, Procopio AD, Albertini MC, Bonafè M, Olivieri F: Anti-senescence compounds: A potential nutraceutical approach to healthy aging. Ageing Res Rev 2018;46:14-31. https://doi.org/10.1016/j.arr.2018.05.001 |
| 84 | Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, Krager K, Ponnappan U, Hauer-Jensen M, Meng A, Zhou D: Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 2016;22:78-83. https://doi.org/10.1038/nm.4010 |
| 85 | Latorre E, Birar VC, Sheerin AN, Jeynes JCC, Hooper A, Dawe HR, Melzer D, Cox LS, Faragher RGA, Ostler EL, Harries LW: Small molecule modulation of splicing factor expression is associated with rescue from cellular senescence. BMC Cell Biol 2017;18:31. https://doi.org/10.1186/s12860-017-0147-7 |
| 86 | McHugh D, Gil J: Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol 2018;217:65-77. https://doi.org/10.1083/jcb.201708092 |
| 87 | Childs BG, Gluscevic M, Baker DJ, Laberge RM, Marquess D, Dananberg J, van Deursen JM: Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov 2017;16:718-735. https://doi.org/10.1038/nrd.2017.116 |
| 88 | de Keizer PL: The fountain of youth by targeting senescent cells? Trends Mol Med 2017;23:6-17. https://doi.org/10.1016/j.molmed.2016.11.006 |
| 89 | Soto-Gamez A, Demaria M: Therapeutic interventions for aging: the case of cellular senescence. Drug Discov Today 2017;22:786-795. https://doi.org/10.1016/j.drudis.2017.01.004 |
| 90 | Burton DGA, Stolzing A: Cellular senescence: Immunosurveillance and future immunotherapy. Ageing Res Rev 2018;43:17-25. https://doi.org/10.1016/j.arr.2018.02.001 |
| 91 | El Ouaamari A, Dirice E, Gedeon N, Hu J, Zhou JY, Shirakawa J, Hou L, Goodman J, Karampelias C, Qiang G, Boucher J, Martinez R, Gritsenko MA, De Jesus DF, Kahraman S, Bhatt S, Smith RD, Beer HD, Jungtrakoon P, Gong Y, Goldfine AB, Liew CW, Doria A, Andersson O, Qian WJ, Remold-O'Donnell E, Kulkarni RN: Serpin B1 promotes pancreatic β cell proliferation. Cell Metab 2016;23:194-205. https://doi.org/10.1016/j.cmet.2015.12.001 |
| 92 | Brüning JC, Winnay J, Bonner-Weir S, Taylor SI, Accili D, Kahn CR: Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell 1997;88:561-572. https://doi.org/10.1016/S0092-8674(00)81896-6 |
| 93 | Kushner JA: The role of aging upon β cell turnover. J Clin Invest 2013;123:990-995. https://doi.org/10.1172/JCI64095 |
| 94 | Bitto A, Ito TK, Pineda VV, LeTexier NJ, Huang HZ, Sutlief E, Tung H, Vizzini N, Chen B, Smith K, Meza D, Yajima M, Beyer RP, Kerr KF, Davis DJ, Gillespie CH, Snyder JM, Treuting PM, Kaeberlein M: Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife 2016;5:e16351. https://doi.org/10.7554/eLife.16351 |
| 95 | Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, O'Hara SP, LaRusso NF, Miller JD, Roos CM, Verzosa GC, LeBrasseur NK, Wren JD, Farr JN, Khosla S, Stout MB, McGowan SJ, Fuhrmann-Stroissnigg H, Gurkar AU, Zhao J, Colangelo D, Dorronsoro A, Ling YY, Barghouthy AS, Navarro DC, Sano T, Robbins PD, Niedernhofer LJ, Kirkland JL: The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 2015;14:644-658. https://doi.org/10.1111/acel.12344 |
| 96 | Lei XG, Vatamaniuk MZ: Two tales of antioxidant enzymes on β cells and diabetes. Antioxid Redox Signal 2011;14:489-503. https://doi.org/10.1089/ars.2010.3416 |
| 97 | Miki A, Ricordi C, Sakuma Y, Yamamoto T, Misawa R, Mita A, Molano RD, Vaziri ND, Pileggi A, Ichii H: Divergent antioxidant capacity of human islet cell subsets: A potential cause of beta-cell vulnerability in diabetes and islet transplantation. PLoS One 2018;13:e0196570. https://doi.org/10.1371/journal.pone.0196570 |
| 98 | Malaisse WJ, Malaisse-Lagae F, Sener A, Pipeleers DG: Determinants of the selective toxicity of alloxan to the pancreatic B cell. Proc Natl Acad Sci U S A 1982;79:927-930. https://doi.org/10.1073/pnas.79.3.927 |
| 99 | Novoselova EG, Glushkova OV, Parfenuyk SB, Khrenov MO, Lunin SM, Novoselova TV, Sharapov MG, Shaev IA, Novoselov VI: Protective effect of peroxiredoxin 6 against toxic effects of glucose and cytokines in pancreatic RIN-m5F β-cells. Biochemistry (Mosc) 2019;84:637-643. https://doi.org/10.1134/S0006297919060063 |
| 100 | Novoselova EG, Glushkova OV, Lunin SM, Khrenov MO, Parfenyuk SB, Novoselova TV, Sharapov MG, Novoselov VI, Fesenko EE: Peroxiredoxin 6 attenuates alloxan-induced type 1 diabetes mellitus in mice and cytokine-induced cytotoxicity in rin-m5f beta cells. J Diabetes Res 2020;2020:7523892. https://doi.org/10.1155/2020/7523892 |
| 101 | Novoselova EG, Glushkova OV, Lunin SM, Khrenov MO, Parfenyuk SB, Novoselova TV, Sharapov MG, Gordeeva AE, Novoselov VI, Fesenko EE: Thymulin and peroxiredoxin 6 have protective effects against streptozotocin-induced type 1 diabetes in mice. Int J Immunopathol Pharmacol 2021;35:20587384211005645. https://doi.org/10.1177/20587384211005645 |
| 102 | Novoselova EG, Glushkova OV, Khrenov MO, Lunin SM, Novoselova TV, Parfenuyk SB: Role of innate immunity and oxidative stress in the development of type 1 diabetes mellitus. Peroxiredoxin 6 as a new anti-diabetic agent. Biochemistry (Mosc) 2021;86:1579-1589. https://doi.org/10.1134/S0006297921120075 |
| 103 | Pacifici F, Arriga R, Sorice GP, Capuani B, Scioli MG, Pastore D, Donadel G, Bellia A, Caratelli S, Coppola A, Ferrelli F, Federici M, Sconocchia G, Tesauro M, Sbraccia P, Della-Morte D, Giaccari A, Orlandi A, Lauro D: Peroxiredoxin 6, a novel player in the pathogenesis of diabetes. Diabetes 2014;63:3210-3220. https://doi.org/10.2337/db14-0144 |
| 104 | Arriga R, Pacifici F, Capuani B, Coppola A, Orlandi A, Scioli MG, Pastore D, Andreadi A, Sbraccia P, Tesauro M, Daniele ND, Sconocchia G, Donadel G, Bellia A, Della-Morte D, Lauro D: Peroxiredoxin 6 is a key antioxidant enzyme in modulating the link between glycemic and lipogenic metabolism. Oxid Med Cell Longev 2019;2019:9685607. https://doi.org/10.1155/2019/9685607 |
| 105 | Novoselova EG, Sharapov MG, Lunin SM, Parfenyuk SB, Khrenov MO, Mubarakshina EK, Kuzekova AA, Novoselova TV, Goncharov RG, Glushkova OV: Peroxiredoxin 6 Applied after exposure attenuates damaging effects of x-ray radiation in 3T3 mouse fibroblasts. Antioxidants (Basel) 2021;10:1951. https://doi.org/10.3390/antiox10121951 |
| 106 | Murakami T, Inagaki N, Kondoh H: Cellular senescence in diabetes mellitus: distinct senotherapeutic strategies for adipose tissue and pancreatic b cells. Front Endocrinol 2022;13:869414. https://doi.org/10.3389/fendo.2022.869414 |
| 107 | Rhodes CJ: Type 2 diabetes-a matter of beta-cell life and death? Science 2005;307:380-384. https://doi.org/10.1126/science.1104345 |
| 108 | Childs BG, Durik M, Baker DJ, van Deursen JM: Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 2015;21:1424-1435. https://doi.org/10.1038/nm.4000 |
| 109 | Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, Gil J, Hara E, Krizhanovsky V, Jurk D, Maier AB, Narita M, Niedernhofer L, Passos JF, Robbins PD, Schmitt CA, Sedivy J, Vougas K, von Zglinicki T, Zhou D, Serrano M, Demaria M: Cellular senescence: defining a path forward. Cell 2019;179:813-827. https://doi.org/10.1016/j.cell.2019.10.005 |
| 110 | Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, Iken M, Vucur M, Weiss S, Heikenwalder M, Khan S, Gil J, Bruder D, Manns M, Schirmacher P, Tacke F, Ott M, Luedde T, Longerich T, Kubicka S, Zender L: Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011;479:547-551. https://doi.org/10.1038/nature10599 |
| 111 | Grosse L, Wagner N, Emelyanov A, Molina C, Lacas-Gervais S, Wagner KD, Bulavin DV: Defined p16(high) senescent cell types are indispensable for mouse healthspan. Cell Metab 2020;32:87-99. https://doi.org/10.1016/j.cmet.2020.05.002 |
| 112 | Midha A., Pan H, Abarca C, Andle J, Carapeto P, Bonner-Weir S, Aguayo-Mazzucato C: Unique human and mouse β-cell senescence-associated secretory phenotype (sasp) reveal conserved signaling pathways and heterogeneous factors. Diabetes 2021;70:1098-1116. https://doi.org/10.2337/db20-0553 |
| 113 | He G, Yan Z, Sun L, Lv Y, Guo W, Gang X, Wang G: Diabetes mellitus coexisted with progeria: a case report of atypical Werner syndrome with novel LMNA mutations and literature review. Endocr J 2019;66:961-969. https://doi.org/10.1507/endocrj.EJ19-0014 |
| 114 | Donadille B, D'Anella P, Auclair M, Uhrhammer N, Sorel M, Grigorescu R, Ouzounian S, Cambonie G, Boulot P, Laforêt P, Carbonne B, Christin-Maitre S, Bignon Y-J, Vigouroux C: Partial lipodystrophy with severe insulin resistance and adult progeria Werner syndrome. Orphanet J Rare Dis 2013;8:106. https://doi.org/10.1186/1750-1172-8-106 |