The Impact of Proinflammatory Cytokines and Imiquimod on GLUT1 in HaCaT Keratinocytes – a Potential Anti-Psoriatic Therapeutic Target?
bFaculty of Biotechnology, University of Wroclaw, Wroclaw, Poland,
cBioimaging LaboratoryŁukasiewiczResearch Network - PORT Polish Center for Technology Development, Wroclaw, Poland
Keywords
Abstract
Background/Aims:
Glucose metabolism has been proven as an essential process for proliferating keratinocytes, which highlights the importance of glucose transporter-1 (GLUT1) not only in the onset of psoriasis but also in the progression and severity of this inflammation-driven disease. In this study, we attempted to find a connection between proinflammatory cytokines (IL-6, IL-17, IL-23, IL-36, TNF-α), a skin inflammation inducing agent – imiquimod (IMQ) and GLUT1 expression.Methods:
Human keratinocyte HaCaT cell line was incubated with exogenous cytokines: IL-6, IL-17A, IL-23, IL-36, TNF-α at a final concentration of 100 ng/ml, or with 1 µM of IMQ, for 48 h. Following the stimulation, glucose uptake and GLUT1 expression were evaluated. The activity of GLUT1 was measured in the presence of a selective GLUT1 inhibitor, BAY-876. The expression of GLUT1 was examined by immunofluorescence and quantified by qPCR, Western blotting and densitometry.Results:
The results from qPCR analysis showed that the administration of exogenous IL-6, IL-17, IL-23 and IL-36 to HaCaT cells resulted in upregulation of GLUT1-encoding SLC2A1 gene, while TNF-α had no significant effect. The same results were confirmed by immunofluorescence analysis, as the fluorescent intensity of GLUT1 was elevated following cytokine and IMQ stimulation. Western blot and densitometry showed that all examined cytokines, as well as IMQ, increased GLUT1 expression. HaCaT cells displayed an improved intracellular 2-deoxy-D-glucose (2-DG) uptake and GLUT1 activity after stimulation by exogenous cytokines and IMQ. The highest uptake of 2-DG was observed after IL-23 stimulation (1.93x) and the lowest after TNF-α stimulation (1.07x). BAY-876 inhibited the 2-DG uptake compared to control.Conclusion:
Our findings suggest that cytokines and IMQ may play a key role in regulating GLUT1 expression in HaCaT cells. We believe that GLUT1 overexpression could potentially be utilized in the targeted treatment of psoriasis.Introduction
Psoriasis is an immune-mediated skin disease manifested by skin inflammation [1, 2]. Typical clinical signs of the disease include increased incidence of plaques and scales, which initiate associating comorbidities such as pain or itch [3]. Similarly to other chronic proinflammatory diseases of the cutaneous system, psoriasis can have a significant impact on both physical and mental health quality [4]. The prevalence of this disease varies from 0.27% to 11.40% and affects approximately 55.8 million adults around the world [5, 6].
The hallmark of psoriasis is constant inflammation that leads to keratinocyte hyperproliferation and disordered differentiation. The early phase of the pathogenesis of psoriasis consists of the activation of autoreactive T cells, which secrete a wide variety of cytokines that play a key role in the development of inflammation. Additionally, various triggers, including Toll-like receptor (TLR) agonists and autoantigens may contribute to the activation of the pathogenic cascade resulting in enhanced production of proinflammatory and proliferation-inducing mediators such as IL-6, IL-17, IL-22, IL-23, IL-36 and TNF-α by immune cells [7, 8]. Among these important cytokines lie potential therapeutic targets for the treatment of psoriasis.
Current treatments for psoriasis seek to minimize inflammation and remove scales [9]. Therapeutic guidelines include topical treatments (mainly corticosteroids, retinoids, vitamin D analogues), phototherapy (UVA and UVB), systemic treatments (immunosuppressants) and systemic biologic treatments [10, 11]. Biopharmaceuticals target specific immune cells that are responsible for psoriasis. The FDA-approved biologic therapies include TNF-α inhibitors (Etanercept , Infliximab , Adalimumab , Certolizumab ), IL-17 inhibitors (Secukinumab , Ixekizumab , Brodalumab ) and IL-23 inhibitors (Guselkumab , Tildrakizumab , Risankizumab ) [12, 13]. There is currently no FDA-approved IL-6 inhibitor therapy for psoriasis, but clinical data have shown the potential [12, 13]. Limited long-term outcome data show that biologics are safe for prolonged use and well-tolerated; however, side effects such as infections, malignancies, cardiac disorders, hepatotoxicity and nerve demyelination have been reported [14]. Thus, novel therapeutic approaches are required to minimize the adverse effects of currently applied therapies.
Glucose is the primary source of energy for all cells, and it is involved in every metabolic cycle and pathway. Due to the high metabolic activity of rapidly proliferating cells in psoriasis, the glucose uptake facilitated by glucose transporters (SGLTs and GLUT) is elevated [15, 16]. A study conducted by Zhang et al. showed that glucose metabolism is crucial for proliferating keratinocytes [17]. Moreover, the genetic deletion of GLUT1 ameliorated psoriasiform hyperplasia induced by imiquimod (IMQ) and IL-23 [17]. Other studies have also found that increased GLUT1 expression in psoriasis lesions promotes keratinocyte proliferation [18] and causes elevated epidermal hyperproliferation, inflammation, and angiogenesis [19]. Interestingly, overexpressed GLUT1 transporters in hyperproliferative keratinocytes may lead to enhanced uptake of selective glucose-conjugated pharmaceuticals [20].
The above-mentioned studies have highlighted GLUT1 as a potential therapeutic target for pathologic hyperproliferation, however, we have failed to find studies examining the impact of cytokines and IMQ on GLUT1 expression and function in psoriasis in vitro . The aim of our study was to find a connection between proinflammatory cytokines (IL-6, IL-17, IL-23, IL-36, TNF-α), IMQ and glucose transporter’s (GLUT1) activity.
Materials and Methods
Cell culture
The HaCaT cell line was obtained from Cell Lines Service (CLS Cell Lines Service, Germany; 300493) and maintained in Dulbecco’s modified Eagle’s medium (DMEM, high glucose, no glutamine) supplemented with 10% v/v fetal bovine serum (FBS), 1% v/v antibiotics (100 U/mL penicillin, 100 µg/mL streptomycin), 1% v/v L-Glutamine at 37°C and 5% CO2 in a humidified incubator. Cell culture reagents were purchased from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). Cells were passaged at 80% confluence. The culture medium was renewed every 3 days. For all experiments, HaCaT cells were incubated with exogenous cytokines: IL-6, IL-17A, IL-23, IL-36, TNF-α (all from Peprotech, USA) for 48 h at a final concentration of 100 ng/ml, or 1 µM imiquimod (IMQ, InVivoGene, USA) for 48 h [21].
RNA extraction and quantitative real-time RT-PCR
The total RNA from cells was isolated using GeneMATRIX Universal RNA Purification Kit (EURx, Gdansk, Poland) according to the manufacturer’s protocol. For first-strand cDNA synthesis we used 1 μg of RNA and then qPCR with SG qPCR Master Mix (EURx) from smART RT-qPCR Kit (EURx) was performed following the manufacturer’s instructions, using Light Cycler 480 instrument (Roche, Basel, Switzerland). The glyceraldehyde 3-phosphate dehydrogenase (G3PDH ) and β-actin (ACTB ) genes were used as housekeeping gene (HKG) standards for GLUT1 gene (SLC2A1 ) expression. Each sample was duplicated, the mean value was used for calculations. Primers for the human SLC2A1 target gene and HGKs were designed using ProbeFinder 2.48 (Roche). The synthesized (Genomed, Poland) forward and reverse sequences of target and HKGs are listed in Table 1.
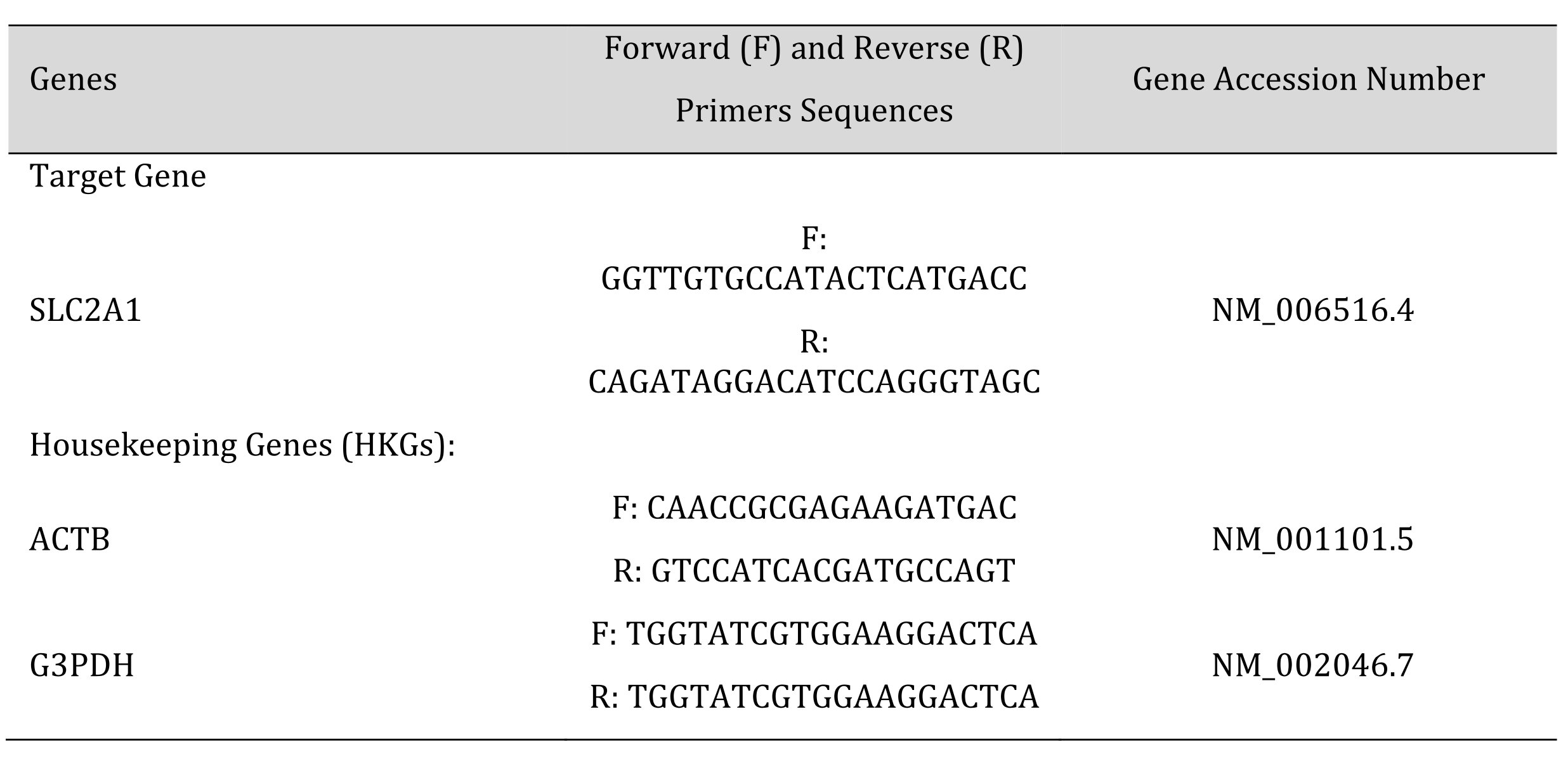
Table 1: DN number at different periods after septoplasty simulation in rats hippocampal formation by staining sections with Nissl toluidine blue stain
Glucose uptake assay
HaCaT cells were seeded in a 96-well culture plate (15x10^3 per well). The next day, cells were stimulated with 100 ng/ml of exogenous cytokines or 1 µM IMQ. After 48 h, cells were washed 3x with phosphate-buffered saline (PBS) and treated with 1 µM BAY-876 [22] (Selleckchem, Houston, TX, USA) and 1 µM insulin [23] (Thermo Fisher Scientific), dissolved in serum-free culture medium, for 2 h. Next, 2-Deoxy-D-glucose (2-DG) (ab136955, Abcam) was added for 20 min and cells were washed 3x with PBS. Cells in each well were lysed with Extraction Buffer from the Glucose Uptake Assay Kit (Colorimetric) (ab136955, Abcam, Cambridge, UK). Further steps were performed according to the manufacturer’s instruction. Samples were diluted by adding 45 μl Assay Buffer to 5 μl of sample. Absorbance (OD) was measured using a microplate reader (Biotek, Winooski, VT, USA) at 412 nm wavelength in a kinetic mode.
Immunofluorescence
For fluorescence confocal microscopy, HaCaT cells were seeded in a 96-well culture plate at 1x10^6 cells/well and after reaching 70% confluence underwent fixation using 4% paraformaldehyde (4% PFA) in PBS for 10 min at room temperature (RT) followed by rinsing (3xPBS) for 3 min (the scheme of rinsing was applied in all steps). After that, a blocking step was performed using a blocking solution (BS) containing 3% Bovine Serum Albumin (Sigma-Aldrich, Saint Louis, MO, USA), 5% Normal Donkey Serum (Abcam), 0.01% Triton X-100 (Sigma-Aldrich), 0.01% Tween 20 (Sigma-Aldrich), 0.3 μM glycine in PBS for 1 h at 4°C. Next, cells were incubated with a primary rabbit anti-human GLUT1 antibody (1:200, clone 16D21, monoclonal, Sigma-Aldrich) diluted in BS overnight at 4°C. The next day, cells were rinsed and incubated with a donkey anti-rabbit DyLight 488 antibody (dilution 1:500, Novus Biologicals, USA) for 2 h at RT in dark conditions. The negative controls were prepared with the omission of the primary antibody. After rinsing, cells were additionally incubated with a phalloidin-Alexa Fluor 555 conjugate (Cytotek, USA) at 37°C for 45 min on a plate rotor for the detection of cellular distribution of actin filaments. Moreover, for the nucleus counterstaining, cells were incubated with 100 μl of PBS containing DAPI (1:10000, Sigma-Aldrich).
Confocal microscopy and image processing
The imaging was performed on a spinning-disk confocal microscope (Carl Zeiss, Oberkochen, Germany) equipped with a dry 20x objective (NA 0.4) and a QImaging Rolera EM-C2 EM-CCD camera. The laser wavelengths used for excitations were 405 nm for DAPI, 488 nm for DyLight 488 (GLUT1), and 561 nm for Alexa Fluor 555 (phalloidin). Emission filters were as follows: BP 450/50 (DAPI), FE01-520/35 (DyLight 488) and BP 600/52 (Alexa Fluor 555). Each condition was imaged in triplicates, and five randomly selected areas per well were analyzed for GLUT1 protein expression. Maximum intensity projections were created from confocal Z-stacks and exported to TIFF files (Zen Microscopy Software, Zeiss). Further steps of fluorescence intensity (FI) analysis were performed in Fiji-ImageJ software (National Institute of Health, Bethesda, USA). In all the channels, Subtract Background and Median filter algorithms were applied to reduce background noise, and images were converted to the 8-bit grey scale. Next, the Huang Threshold algorithm was used on the phalloidin channel to obtain a binary image showing cell bodies, followed by the Watershed algorithm to separate joint objects. Cells were detected using the Analyze particles function and identified regions of interest were transferred onto the GLUT1 channel in order to calculate the FI within them.
Western blot analysis
Following the cell lysis, protein samples (30 μg) were analyzed on 10% SDS-PAGE under non-reducing conditions. Gels were blotted onto a nitrocellulose membrane and transferred at 70 V for 2 h. The membrane was blocked with 5% non-fat dried milk in PBS with 0.1% Tween 20 (Merck Life Science, MI, Italy) for 1 h at RT. Target proteins were detected with the following primary antibodies: anti-GLUT1 polyclonal rabbit IgG (diluted 1:250, Merck, New Jersey, USA) and anti-β-actin monoclonal mouse IgG (diluted 1:5000, Merck). The nitrocellulose membrane was incubated at 4°C overnight with the primary antibody, then 3x washed in PBS with 0.1% Tween 20. Following the washing, the membrane was incubated for 1 h with a secondary anti-rabbit IgG (diluted 1:10000, Merck) conjugated to horseradish peroxidase. Western blot bands were detected with Super Signal® West PICO (Thermo Fisher Scientific) and visualized with ChemiDOC XRS, Quantity One 4.6.5 software (Bio-Rad Laboratories, Segrate, Milano, Italy). Western blot semi quantitative calculations were prepared using ImageJ software (version 2.1.4.7 i1).
Statistical analysis
All experiments were conducted in triplicates. Data were presented as means ± SD (as stated in figure legends). Statistical significance was determined by ANOVA with post-hoc Holm-Sidak’s multiple comparisons test using GraphPad Prism 9 software. p < 0.05 was considered as statistically significant.
Results
The expression of GLUT1 is significantly induced by IL-6, IL-17, IL-23, IL-36 and IMQ in HaCaT keratinocytes
To investigate a potential connection between proinflammatory cytokines, IMQ and glucose metabolism in keratinocytes, we examined whether GLUT1 expression is altered upon IL-6, IL-17, IL-23, IL-36, TNF-α and IMQ stimulation. We treated HaCaT cells with exogenous cytokines and IMQ and analyzed their impact on GLUT1 expression. Briefly, RNA from control and treated keratinocytes were reverse-transcribed into cDNA and analyzed by qPCR using primers specific to GLUT1. The results showed that the administration of exogenous IL-6, IL-17, IL-23 and IL-36 to HaCaT cells resulted in the upregulation ofthe SLC2A1 gene, while TNF-α had no significant effect (Fig. 1). A statistical difference in the SLC2A1 mRNA expression level was also found between IMQ-treated cells and control. These observations demonstrate that GLUT1, which is the primary glucose transporter expressed in keratinocytes, is upregulated upon cytokine and IMQ-induced inflammation.
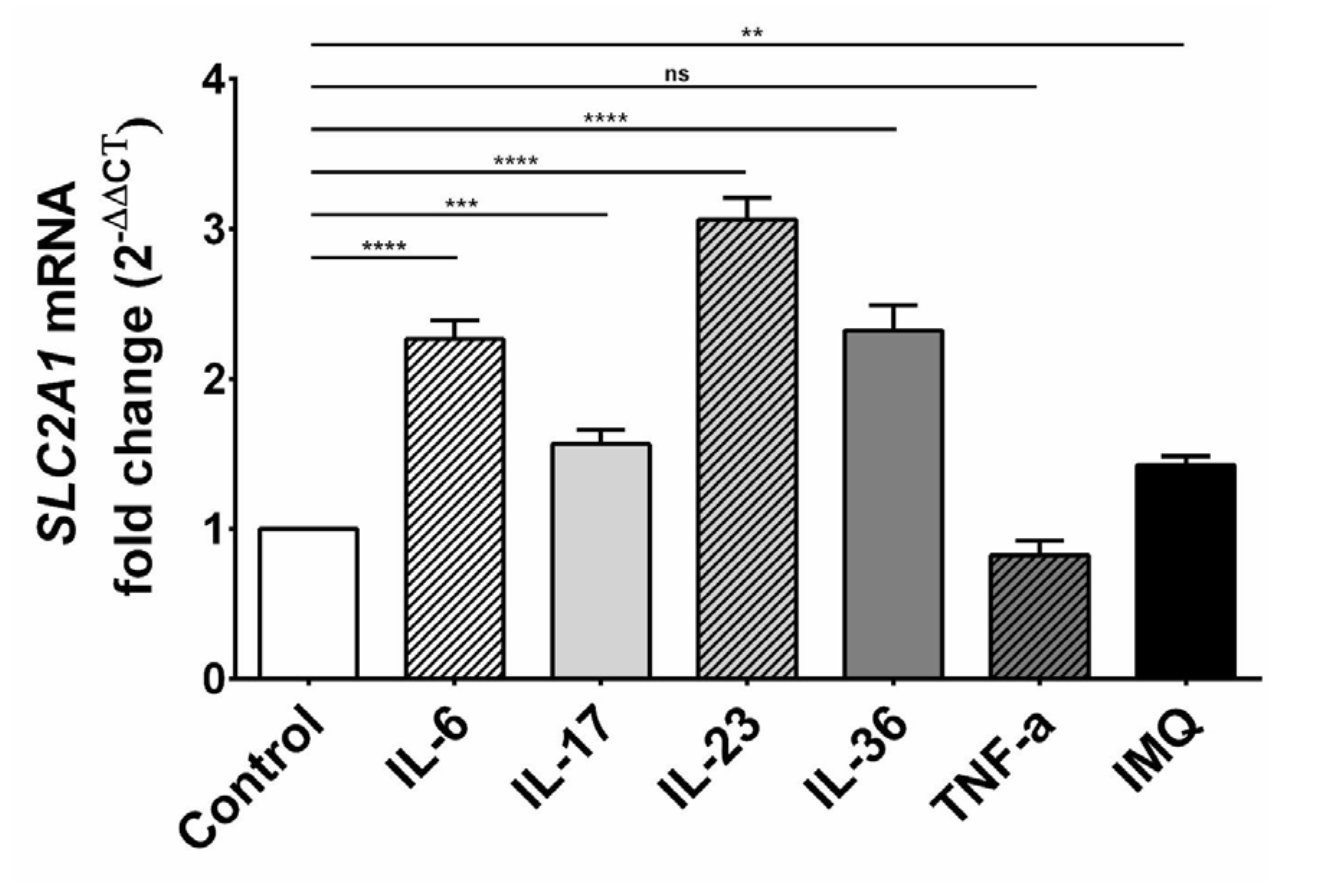
Fig. 1: Expression level of SLC2A1 mRNA in HaCaT cells after stimulation with exogenous cytokines and IMQ. The statistical significance level was set at p = 0.01-0.05 (*), 0.001-0.01 (**), 0.0001-0.001 (***), p ≤ 0.0001 (****), p > 0.05 = not significant (ns).
Incubation of HaCaT cells with exogenous cytokines and IMQ increases the intracellular uptake of glucose
To evaluate the effect of proinflammatory cytokines and IMQ on the glucose uptake by HaCaT cells, we measured the concentration of 2-DG in the intracellular compartment by using the Glucose Uptake Assay. Our results showed enhanced intracellular glucose uptake and GLUT1 activity in HaCaT cells stimulated by exogenous cytokines and IMQ. The highest uptake of 2-DG was observed after IL-23 stimulation (1.93x) and lowest after TNF-α stimulation (1.07x). 2-DG concentration was increased 1.47x for IL-6, 1.44x for IL-17, 1.62x for IL-36 and 1.6x for IMQ (Fig. 2). Additionally, the stimulation of cells with a selective GLUT1 inhibitor (BAY-876) led to a decrease of 2-DG uptake compared to control (Fig. 2). These results demonstrate that IMQ and proinflammatory cytokines, except for TNF-α, significantly regulate the intracellular glucose uptake and metabolism during the inflammation.
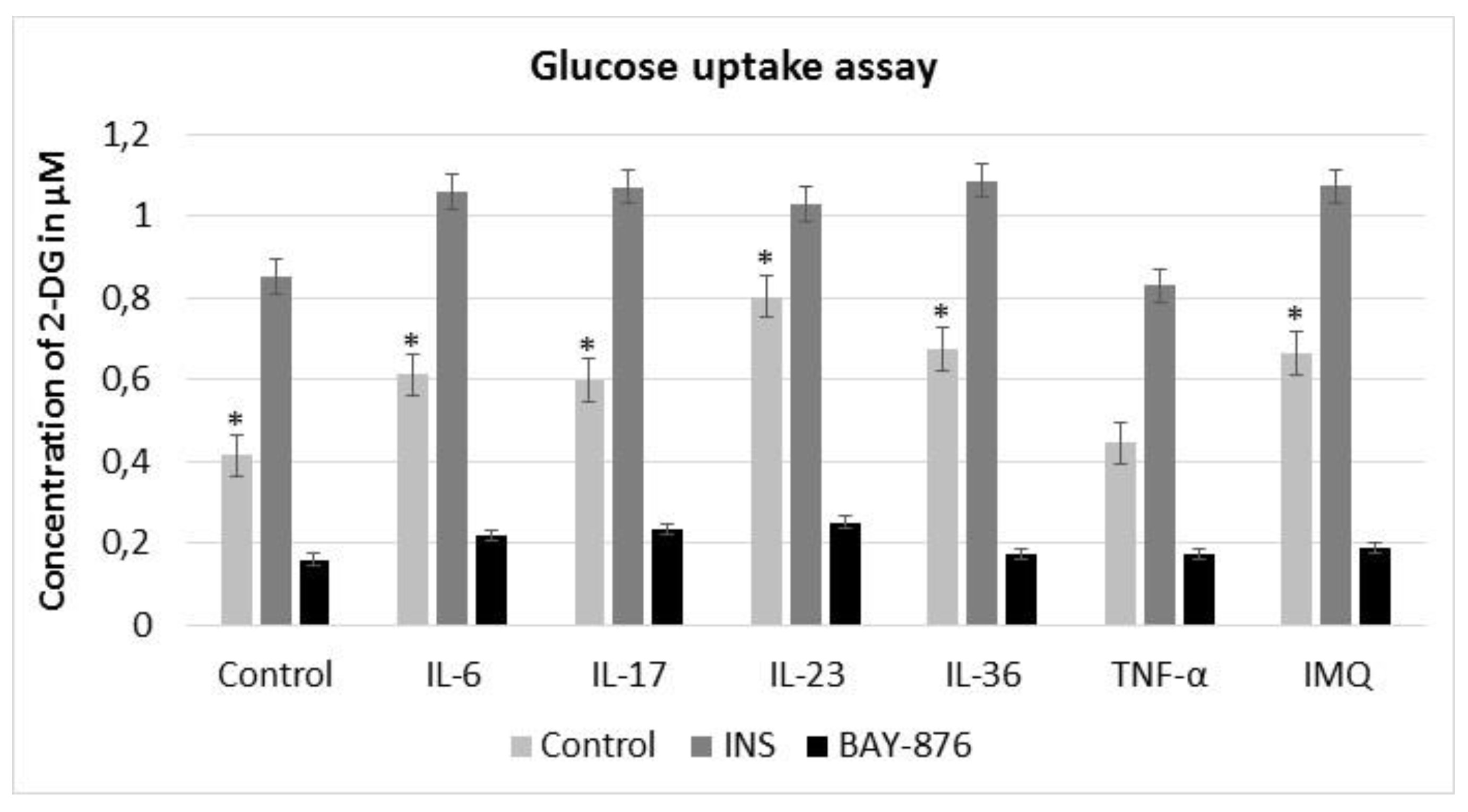
Fig. 2: 2-DG uptake in HaCaT cells after stimulation with insulin (INS), BAY-876 inhibitor, exogenous cytokines and IMQ. Insulin significantly increased the uptake of 2-DG, while GLUT1 selective inhibitor BAY-876 decreased the concentration of 2-DG in HaCaT cells. The intracellular glucose uptake and GLUT1 activity in HaCaT cells stimulated with IMQ and proinflammatory cytokines was enhanced. The experiment was conducted thrice. Data represent mean D; *p .05, compared to untreated control.
Exogenous cytokines and IMQ increase the fluorescence intensity of GLUT1 in HaCaT cells
To investigate the impact of cytokines and IMQ on the GLUT1 protein level in HaCaT cells, we performed a quantitative analysis of the fluorescence intensity (FI) of the GLUT1 signal following the stimulations. GLUT1 protein localization was visualized using confocal microscopy. The immunostaining revealed that GLUT1 localized primarily to the cell membrane but was also visible in the perinuclear region, which is clearly visible on the overlay with DAPI staining (Fig. 3). Most importantly, the fluorescence intensity of GLUT1 in HaCaT cells was higher after incubation with IL-6, IL-17, IL-23, IL-36 and IMQ, while TNF-α did not significantly affect the GLUT1 level (Fig. 4).
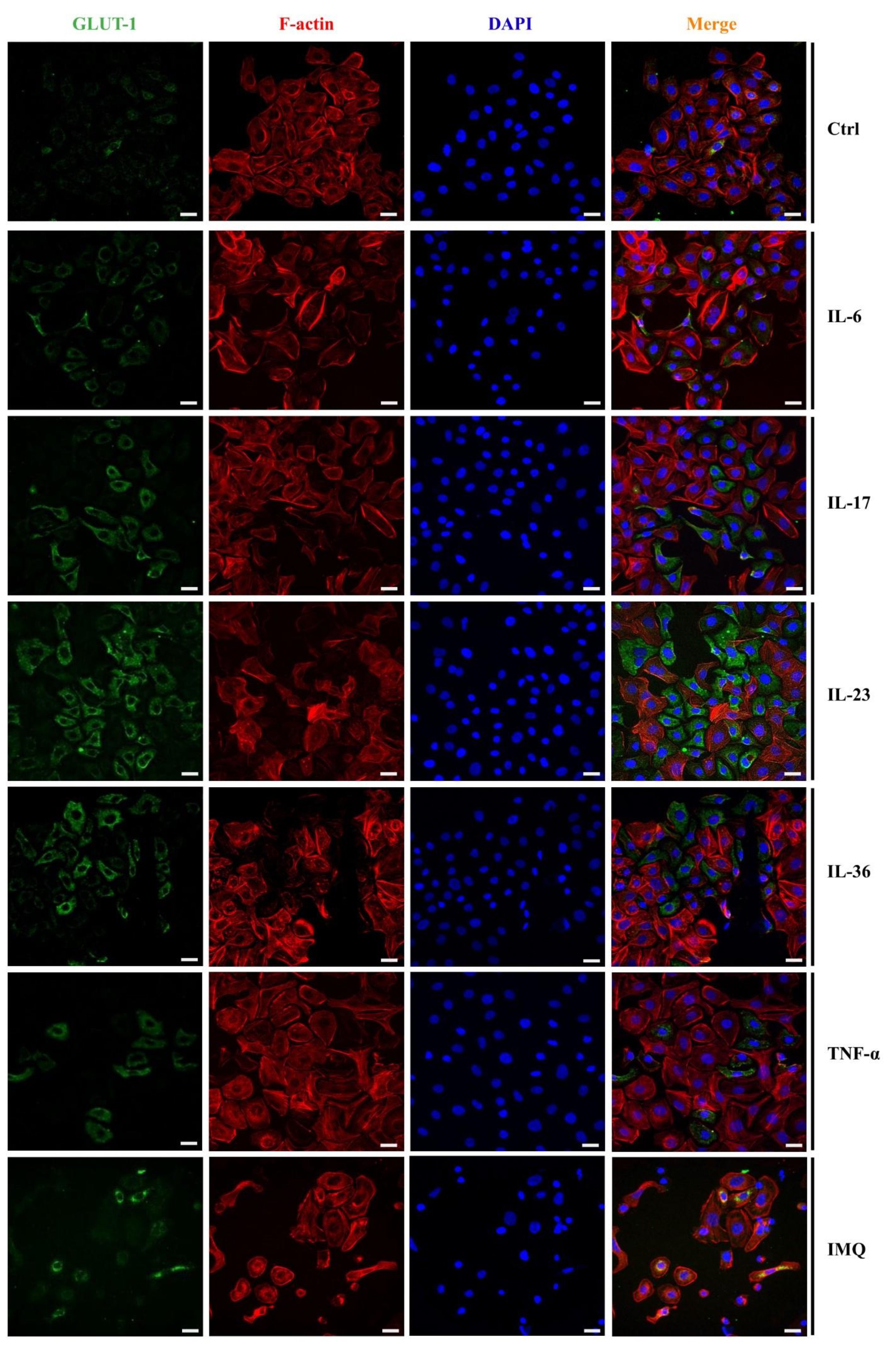
Fig. 3: Immunofluorescence staining of GLUT1 in HaCaT cells after the stimulation with exogenous proinflammatory cytokines and IMQ. Scale bar = 20 μm.
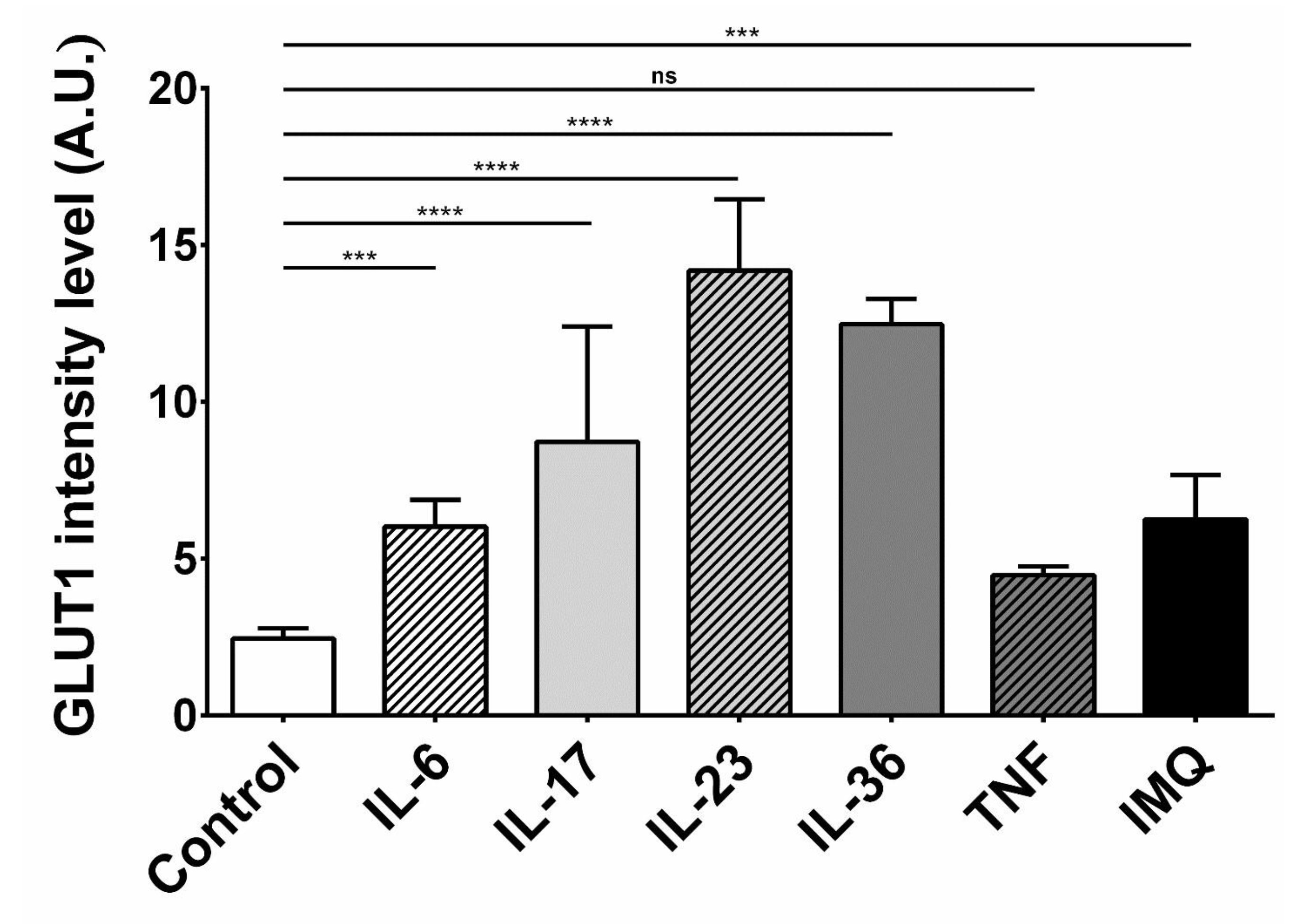
Fig. 4: Fluorescence intensity analysis of the GLUT1 signal in HaCaT cells after stimulation with exogenous cytokines and IMQ. The analysis was performed in the Fiji-ImageJ software. The statistical significance level was set at p = 0.01-0.05 (*), 0.001-0.01 (**), 0.0001-0.001 (***), p ≤ 0.0001 (****), p ≥ 0.05 = ns = not significant.
Western blot and densitometry prove the correlation between proinflammatory cytokines and the increase of GLUT1 expression
Using Western blot method, it was shown that all of the examined cytokines, as well as IMQ, increased the GLUT1 expression (Fig. 5). The relative expression analysis revealed that HaCaT cells, cultured with IL-36 and IMQ expressed almost 80% (p < 0.05) more of GLUT1 protein, compared to control cells cultured in the standard medium, while IL-23 and TNF-α increased GLUT1 levels by ∼40% (p < 0.05). IL-6 and IL-17 treated cells present ∼60% higher relative expression than control cells.
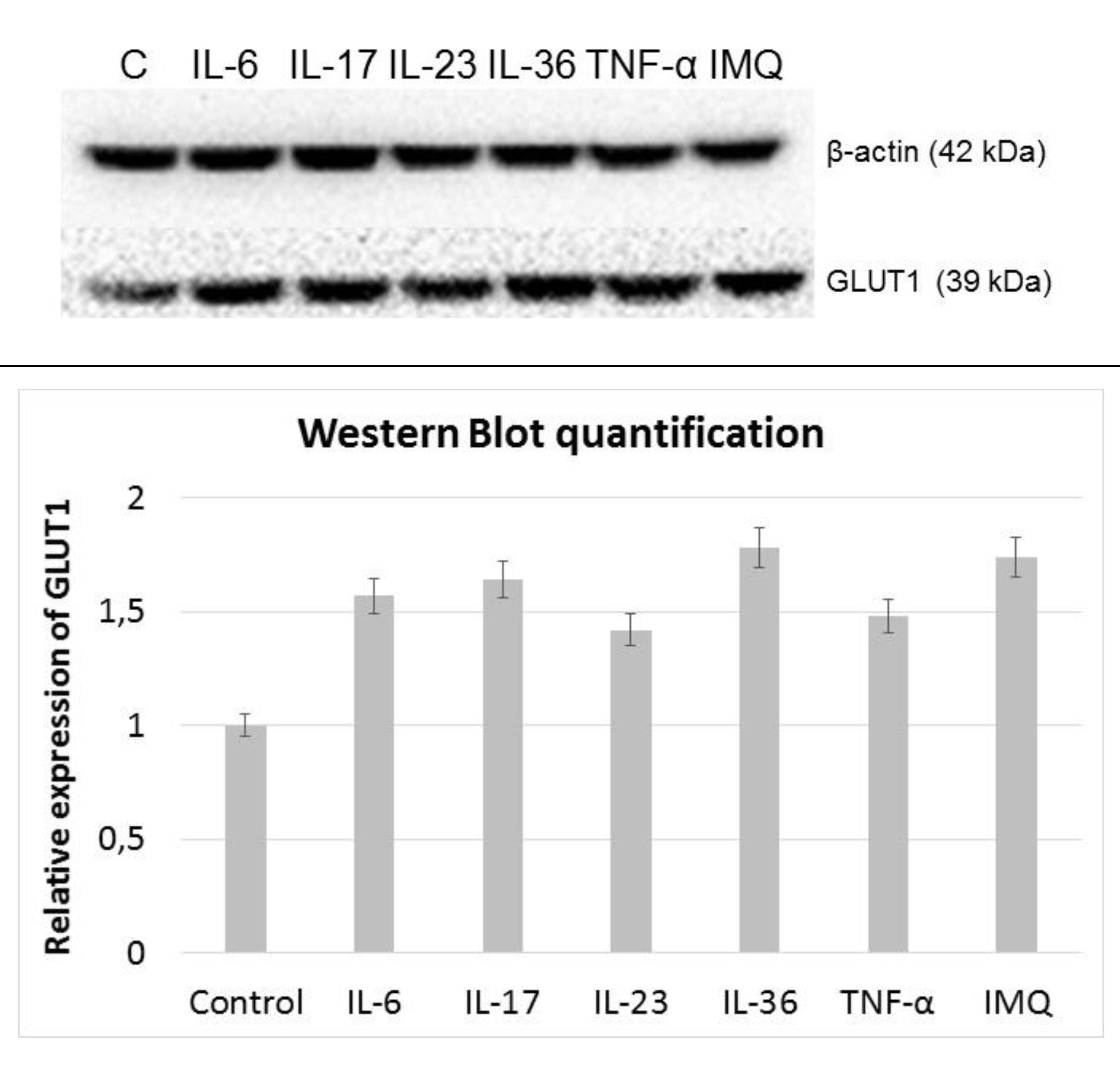
Fig. 5: Western Blot analysis of the GLUT1 signal in HaCaT cells after stimulation with exogenous cytokines and IMQ.
Discussion
Glucose is the major energy source for rapidly proliferating keratinocytes during the psoriasis progression. Its uptake is mediated and precisely regulated by glucose transporters (GLUTs). Among thirteen GLUT family members known to date, only GLUT1 has been identified as a significant hexose transporter expressed in keratinocytes and activated T lymphocytes [17]. In this study, we found that the exogenous administration of pro-inflammatory cytokines (IL-6, IL-17, IL-23, IL-36) and IMQ increases GLUT1 expression and results in elevated glucose uptake in inflamed keratinocytes. Surprisingly, stimulation of cells with TNF-α played a minor role in the expression of GLUT1. The pathogenesis of psoriasis involves precise communication between keratinocytes, immune cells, and other skin-related cells. A variety of different inflammatory cytokines are involved in this process, including IL-4, IL-6, IL-23, IL-17, IL-22, IL-36, and TNF-α. Their increased secretion triggers multiple cell signaling pathways, resulting in excessive keratinocyte proliferation and production of antimicrobial peptides, chemokines, and growth factors contributing to the amplification of inflammation [24–26]. The discovery that cytokines provide signals to promote glucose uptake has already been shown in several studies [27–29], but only few of them have highlighted this phenomenon in the case of psoriasis. Hodeib et al. found that rapidly proliferating keratinocytes express high levels of GLUT1, leading to an increased epidermal thickness, inflammatory cell density, and microvessel density [19]. Furthermore, as suggested by Ebeling et al., the upregulation of GLUT1 expression increases glucose transport, which seems to have an impact on insulin resistance, especially in severe psoriasis cases [30]. The positive correlation between GLUT1 expression and progressive keratinocyte proliferation, shown by increased Ki-67 expression in psoriasis lesions, has also been indicated by Abdou et al. [31]. Consistently with our results, Huang et al. also observed membrane-enriched GLUT1 in keratinocytes from the psoriatic skin [32]. These findings reinforce our suggestions regarding the potential of glucose metabolic-targeted therapy. Furthermore, according to Zhang et al. study, GLUT1 deletion or inhibition abolished glucose transport by 95% in keratinocytes and impaired their proliferation in vitro . However, despite targeted GLUT1 deletion in keratinocytes, there was no apparent difference in skin morphology in vivo , suggesting that alternative substrates may be responsible for complementing the requirement for glucose, such as amino acids, fatty acids, or other hexoses. Moreover, the same study showed that GLUT1 downregulation renders the skin resistant to IMQ- or IL-23-induced psoriasiform hyperplasia [17]. This finding, in accordance with our results, suggests that cytokines and IMQ may play a key role in regulating GLUT1 expression. Taking into account that GLUT1 deletion in keratinocytes dampens psoriasiform hyperplasia and the GLUT1 overexpression is mediated by immune cells, we believe that inflammatory cytokines should be taken into account while precisely designing pharmaceuticals targeting GLUT1. Based on our data, we believe that GLUT1 overexpression could potentially be utilized by using selective glucose-conjugated pharmaceuticals against psoriasis. Thus, we propose a new drug development path that seems to alleviate psoriasis symptoms by their selective glucose-conjugated uptake by cytokine-induced GLUT1 overexpression. However, further studies are required to precisely dissect the utility of GLUT1 in the potential treatment of psoriasis.Acknowledgements
Author Contributions
Sebastian Makuch was responsible for the conceptualization of the study, original draft preparation, conduction of the experiments and manuscript editing. Piotr Kupczyk and Alicja Makarec conducted the experiments and edited the manuscript; Grzegorz Chodaczek analyzed the data, prepared some of the figures and edited the manuscript. Piotr Ziółkowski received the funding and supervised the study. Marta Woźniak edited the manuscript and supervised the study. All authors have read and agreed to the published version of the manuscript.
Funding Sources
This research was funded by the National Science Centre, Poland, grant number 2019/35/O/NZ4/01463
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors have no conflicts of interest to declare.
References
| 1 | Griffiths CE, Barker JN: Pathogenesis and clinical features of psoriasis. Lancet 2007;370:263-271.
https://doi.org/10.1016/S0140-6736(07)61128-3 |
| 2 | Armstrong AW, Read C: Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. JAMA - J Am Med Assoc 2020;323:1945-1960.
https://doi.org/10.1001/jama.2020.4006 |
| 3 | Jaworecka K, Muda-Urban J, Rzepko M, Reich A: Molecular aspects of pruritus pathogenesis in psoriasis. Int J Mol Sci 2021;22:1-11.
https://doi.org/10.3390/ijms22020858 |
| 4 | Wu JJ, Feldman SR, Koo J, Marangell LB: Epidemiology of mental health comorbidity in psoriasis. J Dermatolog Treat 2018;29:487-495.
https://doi.org/10.1080/09546634.2017.1395800 |
| 5 | Parisi R, Symmons DPM, Griffiths CEM, Ashcroft DM: Global epidemiology of psoriasis: A systematic review of incidence and prevalence. J Invest Dermatol 2013;133:377-385.
https://doi.org/10.1038/jid.2012.339 |
| 6 | Parisi R, Iskandar IYK, Kontopantelis E, Augustin M, Griffiths CEM, Ashcroft DM: National, regional, and worldwide epidemiology of psoriasis: Systematic analysis and modelling study. BMJ 2020;369.
https://doi.org/10.1136/bmj.m1590 |
| 7 | Chiricozzi A, Romanelli P, Volpe E, Borsellino G, Romanelli M: Scanning the immunopathogenesis of psoriasis. Int J Mol Sci 2018;19:179.
https://doi.org/10.3390/ijms19010179 |
| 8 | Georgescu SR, Tampa M, Caruntu C, Sarbu MI, Mitran CI, Mitran MI, Matei C, Constantin C, Neagu M: Advances in understanding the immunological pathways in Psoriasis. Int J Mol Sci 2019;20:3:739.
https://doi.org/10.3390/ijms20030739 |
| 9 | Schadler ED, Ortel B, Mehlis SL: Biologics for the primary care physician: Review and treatment of psoriasis. Disease-a-Month 2019;65:51-90.
https://doi.org/10.1016/j.disamonth.2018.06.001 |
| 10 | Menter A, Strober BE, Kaplan DH, Kivelevitch D, Prater EF, Stoff B, Armstrong AW, Connor C, Cordoro KM, Davis DMR, Elewski BE, Gelfand JM, Gordon KB, Gottlieb AB, Kavanaugh A, Kiselica M, Korman NJ, Kroshinsky D, Lebwohl M, Leonardi CL, Lichten J, Lim HW, Mehta NN, Paller AS, Parra SL, Pathy AL, Rupani RN, Siegel M, Wong EB, Wu JJ, Hariharan V, Elmets CA: Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol 2019;80:1029-1072.
https://doi.org/10.1016/j.jaad.2018.11.057 |
| 11 | Elmets CA, Lim HW, Stoff B, Connor C, Cordoro KM, Lebwohl M, Armstrong AW, Davis DMR, Elewski BE, Gelfand JM, Gordon KB, Gottlieb AB, Kaplan DH, Kavanaugh A, Kiselica M, Kivelevitch D, Korman NJ, Kroshinsky D, Leonardi CL, Lichten J, Mehta NN, Paller AS, Parra SL, Pathy AL, Farley Prater EA, Rupani RN, Siegel M, Strober BE, Wong EB, Wu JJ, Hariharan V, Menter A: Joint American Academy of Dermatology-National Psoriasis Foundation guidelines of care for the management and treatment of psoriasis with phototherapy. J Am Acad Dermatol 2019;81:775-804.
https://doi.org/10.1016/j.jaad.2019.04.042 |
| 12 | Choy EH, De Benedetti F, Takeuchi T, Hashizume M, John MR, Kishimoto T: Translating IL-6 biology into effective treatments. Nat Rev Rheumatol 2020;16:335-345.
https://doi.org/10.1038/s41584-020-0419-z |
| 13 | Tsai YC, Tsai TF: Anti-interleukin and interleukin therapies for psoriasis: current evidence and clinical usefulness. Ther Adv Musculoskelet Dis 2017;9:277-294.
https://doi.org/10.1177/1759720X17735756 |
| 14 | Rustin MH: Long-term safety of biologics in the treatment of moderate-to-severe plaque psoriasis: review of current data. Br J Dermatol 2012;167 Suppl:3-11.
https://doi.org/10.1111/j.1365-2133.2012.11208.x |
| 15 | Hiebert P, Werner S: Targeting metabolism to treat psoriasis. Nat Med 2018;24:537-539.
https://doi.org/10.1038/s41591-018-0027-5 |
| 16 | Cibrian D, de la Fuente H, Sánchez-Madrid F: Metabolic Pathways That Control Skin Homeostasis and Inflammation. Trends Mol Med 2020;26:975-986.
https://doi.org/10.1016/j.molmed.2020.04.004 |
| 17 | Zhang Z, Zi Z, Lee EE, Zhao J, Contreras DC, South AP, Abel ED, Chong BF, Vandergriff T, Hosler GA, Scherer PE, Mettlen M, Rathmell JC, Deberardinis RJ, Wang RC: Differential glucose requirement in skin homeostasis and injury identifies a therapeutic target for psoriasis article. Nat Med 2018;24:617-627.
https://doi.org/10.1038/s41591-018-0003-0 |
| 18 | Tao J, Yang J, Wang L, Li Y, Liu YQ, Dong J, Li L, Wen X, Shen GX, Tu YT: Expression of GLUT-1 in psoriasis and the relationship between GLUT-1 upregulation induced by hypoxia and proliferation of keratinocyte growth. J Dermatol Sci 2008;51:203-207.
https://doi.org/10.1016/j.jdermsci.2008.04.012 |
| 19 | Hodeib AAH, Neinaa YMEH, Zakaria SS, Alshenawy HAS: Glucose transporter-1 (GLUT-1) expression in psoriasis: correlation with disease severity. Int J Dermatol 2018;57:943-951.
https://doi.org/10.1111/ijd.14037 |
| 20 | Makuch S, Wozniak M, Krawczyk M, Pastuch-Gawołek G, Szeja W, Agrawal S: Glycoconjugation as a promising treatment strategy for psoriasis. J Pharmacol Exp Ther 2020;373:204-212.
https://doi.org/10.1124/jpet.119.263657 |
| 21 | Cho KA, Kim JY, Woo SY, Park HJ, Lee KH, Pae CU: Interleukin-17 and interleukin-22 induced proinflammatory cytokine production in keratinocytes via inhibitor of nuclear factor-b kinase-a expression. Ann Dermatol 2012;24:398-405.
https://doi.org/10.5021/ad.2012.24.4.398 |
| 22 | Wu Q, Ba-Alawi W, Deblois G, Cruickshank J, Duan S, Lima-Fernandes E, Haight J, Tonekaboni SAM, Fortier AM, Kuasne H, McKee TD, Mahmoud H, Kushida M, Cameron S, Dogan-Artun N, Chen W, Nie Y, Zhang LX, Vellanki RN, Zhou S, Prinos P, Wouters BG, Dirks PB, Done SJ, Park M, Cescon DW, Haibe-Kains B, Lupien M, Arrowsmith CH: GLUT1 inhibition blocks growth of RB1-positive triple negative breast cancer. Nat Commun 2020;11:1-12.
https://doi.org/10.1038/s41467-020-18020-8 |
| 23 | Zhang WY, Lee JJ, Kim Y, Kim IS, Han JH, Lee SG, Ahn MJ, Jung SH, Myung CS: Effect of eriodictyol on glucose uptake and insulin resistance in vitro. J Agric Food Chem 2012;60:7652-7658.
https://doi.org/10.1021/jf300601z |
| 24 | Rendon A, Schäkel K: Psoriasis pathogenesis and treatment. Int J Mol Sci 2019;20:6:1475.
https://doi.org/10.3390/ijms20061475 |
| 25 | Dopytalska K, Ciechanowicz P, Wiszniewski K, Szymańska E, Walecka I: The role of epigenetic factors in psoriasis. Int J Mol Sci 2021;22:17:9294.
https://doi.org/10.3390/ijms22179294 |
| 26 | Zhou X, Chen Y, Cui L, Shi Y, Guo C: Advances in the pathogenesis of psoriasis: from keratinocyte perspective. Cell Death Dis 2022;13:1-13.
https://doi.org/10.1038/s41419-022-04523-3 |
| 27 | Wieman HL, Wofford JA, Rathmell JC: Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell 2007;18:1437-1446.
https://doi.org/10.1091/mbc.e06-07-0593 |
| 28 | Shikhman AR, Brinson DC, Valbracht J, Lotz MK: Cytokine Regulation of Facilitated Glucose Transport in Human Articular Chondrocytes. J Immunol 2001;167:7001-7008.
https://doi.org/10.4049/jimmunol.167.12.7001 |
| 29 | Phillips T, Ferraz I, Bell S, Clegg PD, Carter SD, Mobasheri A: Differential regulation of the GLUT1 and GLUT3 glucose transporters by growth factors and pro-inflammatory cytokines in equine articular chondrocytes. Vet J 2005;169:216-222.
https://doi.org/10.1016/j.tvjl.2004.01.026 |
| 30 | Ebeling P, Koistinen HA, Koivisto VA: Insulin-independent glucose transport regulates insulin sensitivity. FEBS Lett 1998;436:301-303.
https://doi.org/10.1016/S0014-5793(98)01149-1 |
| 31 | Abdou AG, Maraee AH, Eltahmoudy M, El-Aziz RA: Immunohistochemical expression of GLUT-1 and Ki-67 in chronic plaque psoriasis. Am J Dermatopathol 2013;35:731-737.
https://doi.org/10.1097/DAD.0b013e3182819da6 |
| 32 | Huang X, Chen J, Zeng W, Wu X, Chen M, Chen |