Can Iron and Polyunsaturated Fatty Acid Supplementation Induce Ferroptosis?
Keywords
Abstract
Introduction
Ferroptosis was first reported in 2012. It was described as a new form of regulated (non-apoptotic) cell death involving morphological and biochemical alterations that differ from previously identified and characterized types of cell death such as apoptosis, necrosis, and autophagy. Ferroptosis is triggered by excessive iron-induced lipid peroxidation [1].
Ferroptosis involves the inhibition of glutathione peroxidase 4 (GPX4) and a consequent increase in lipid peroxide formation. Under normal physiological conditions, GPX4 converts toxic lipid hydroperoxides (L-OOH) into non toxic lipid alcohols (L-OH) [2]. Ferroptosis is also associated with excess Fe3+ in the intracellular medium. The Fe3+ is then reduced to Fe2+ which catalyzes ferroptosis by increasing reactive oxygen species (ROS) formation by Fenton and Haber Weiss reactions [3]. Thus, GPX4 inhibition increases ROS by allowing excess intracellular iron accumulation and reducing the antioxidant capacity. In this manner, it promotes the peroxidation of polyunsaturated fatty acids (PUFAs) in cell membranes.
Under certain conditions, the consumption of key nutrients, such as iron and PUFAs, might potentially cause ferroptosis. The efficiency of intestinal iron absorption determines the iron status in the body, as well as the opposite also occurs (body iron status influences the efficiency of absorption of this nutrient) [4]. In addition, blood transfusions necessary in the treatment of some diseases may also increase the iron content in the body. Excess iron can also be present in some hereditary diseases, such as hemochromatosis [5]. In this context, it is known that polyunsaturated fatty acids (PUFAs) such as linoleic acid (LA), alpha-linolenic acid (ALA), eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and arachidonic acid (AA) are more susceptible to oxidation. The integration of PUFAs into cell membranes may stimulate ferroptosis in the presence of excess intracellular iron [6]. Hence, in situations of excess iron in the body, PUFA supplementation could hypothetically trigger ferroptosis. At the same time, it is known that the Western diet is rich in omega-6 PUFAs [7], which would also be of concern for individuals with excess iron in the body. In this context, the main concern about PUFA ingestion is for individuals with excess iron in the body (either by supplementation, blood transfusion or genetic diseases). On the other hand, the most likely cellular targets for the occurrence of ferroptosis through iron supplementation may be the intestinal cells themselves and the intestinal microbiota, since, in humans, the intestinal absorption of iron is regulated by means of hepcidin [4].
The aim of the present review was to search and compile evidence from the literature for the effects of dietary iron and PUFA supplementation on ferroptosis. In this sense, the aim is to find out whether it is possible to control the cell death process via ferroptosis by means of food or nutritional supplementation.
Ferroptosis
Ferroptosis is driven by two concomitant processes. In the first, the selenoenzyme phospholipid hydroperoxide glutathione peroxidase 4 (GPX4), which protects cells against lipid peroxidation, is suppressed [3]. In the second, transferrin receptors increase intracellular iron flux [8]. Therefore, lipid peroxidation is exacerbated by the presence of intracellular iron while GPX4 suppression diminishes the detoxification of lipid peroxides generated in response to increasing intracellular ROS content. Lipid peroxides translocate the BH3 interacting-domain death agonist pro-apoptotic protein (BID), increase membrane porosity, attenuate selective permeability, release organelles into the extracellular medium, and cause ferroptosis [9].
Under physiological conditions, GPX4 reduces lipid peroxides to their corresponding lipid alcohols [10]. When GPX4 is suppressed, however, lipid peroxides can accumulate in the cell. GPX4 is composed of eight nucleophilic amino acids including seven cysteines and one selenocysteine. The latter is directly dependent on selenium [10].
GPX4 may be suppressed in different ways. Erastin inhibits the cystine antiporter System Xc- and prevents its entry into the cytosol (Fig. 1). Glutathione synthetase and glutamate cysteine synthase form the tripeptide glutathione - which is a cofactor of GPX4 – and consists of glutamate, glycine, and cysteine [11]. A decrease in cystine results in a reduction in cysteine and the depletion of glutathione. Hence, cystine uptake is essential for GPX4 activity and, by extension, ferroptosis resistance [11] (Fig. 1). GPX4 is also suppressed by chemoproteomics such as lethal selective RAS 3 (RSL-3) which binds selenocysteine at the GPX4 active site and impairs its antioxidant function (Fig. 1) [12]. GPX4 may also be suppressed by ferroptosis 56 inhibitor (FIN56) which promotes GPX4 degradation and by 1, 2-dioxolane-containing endoperoxide (FINO2) which inhibits GPX4, oxidizes iron, and causes generalized lipid peroxidation (Fig. 1) [13]. The foregoing findings suggest that endoperoxides such as FINO2 may initiate ferroptosis through multiple mechanisms [13].
Lipid peroxides are produced by Fe-dependent pathways [3]. They are produced through the Fenton reaction (Fig. 1). The Fe2+ in labile iron reservoirs reacts with hydrogen peroxide in the ambient medium to form hydroxyl radicals (•OH) (Fig. 1). The PUFAs in cell membranes contain multiple double hydrogen bonds that can react with hydroxyl radical, thereby triggering lipid peroxidation (Fig. 1). Lipid peroxides may also be generated by the oxygenation and esterification of PUFAs such as AA [3]. Elongase converts AA to adrenoil (AdA) which, in turn, is a substrate for acyl CoA synthetase family 4 long chain (ACSL4). The latter enzyme oxidizes AdA to arachidonyl-CoA (AA CoA). AA-CoA may also be esterified by lysophosphatidylcholine acyltransferase 3 (LPCAT3) to AA-containing phosphatidylethanolamine (AA-PE). AA-PE is then oxidized by Fe-dependent lipoxygenases to the toxic phospholipid hydroperoxide AA-OOH-PE (Fig. 1). Ferroptosis occurs when the AA-OOH-PE levels exceed the cell threshold (Fig. 1) [3]. The phospholipid phosphatidylethanolamine (PE) containing AA or AdA can cause ferroptosis. Modulation of the genes regulating the fatty acid metabolism mediator acyl-CoA synthetase (ACSL4) and LPCAT3 can cause ferroptosis [12]. ACSL4 preferentially acetylates AA [14] while LPCAT3 catalyzes the integration of acetylated AA into membrane phospholipids [15]. Hence, ACSL4 and LPCAT3 deletion may suppress ferroptosis and limitthe oxidation-sensitive FA pool in cell membranes. Lipid peroxides may also be generated by lipid auto-oxidation. This autocatalytic reaction occurs in a free radical chain that generates lipid hydroperoxides in the presence of iron [3]. GPX4 depletion or inactivation may increase ferroptosis to a greater extent than PUFA oxidation [3].
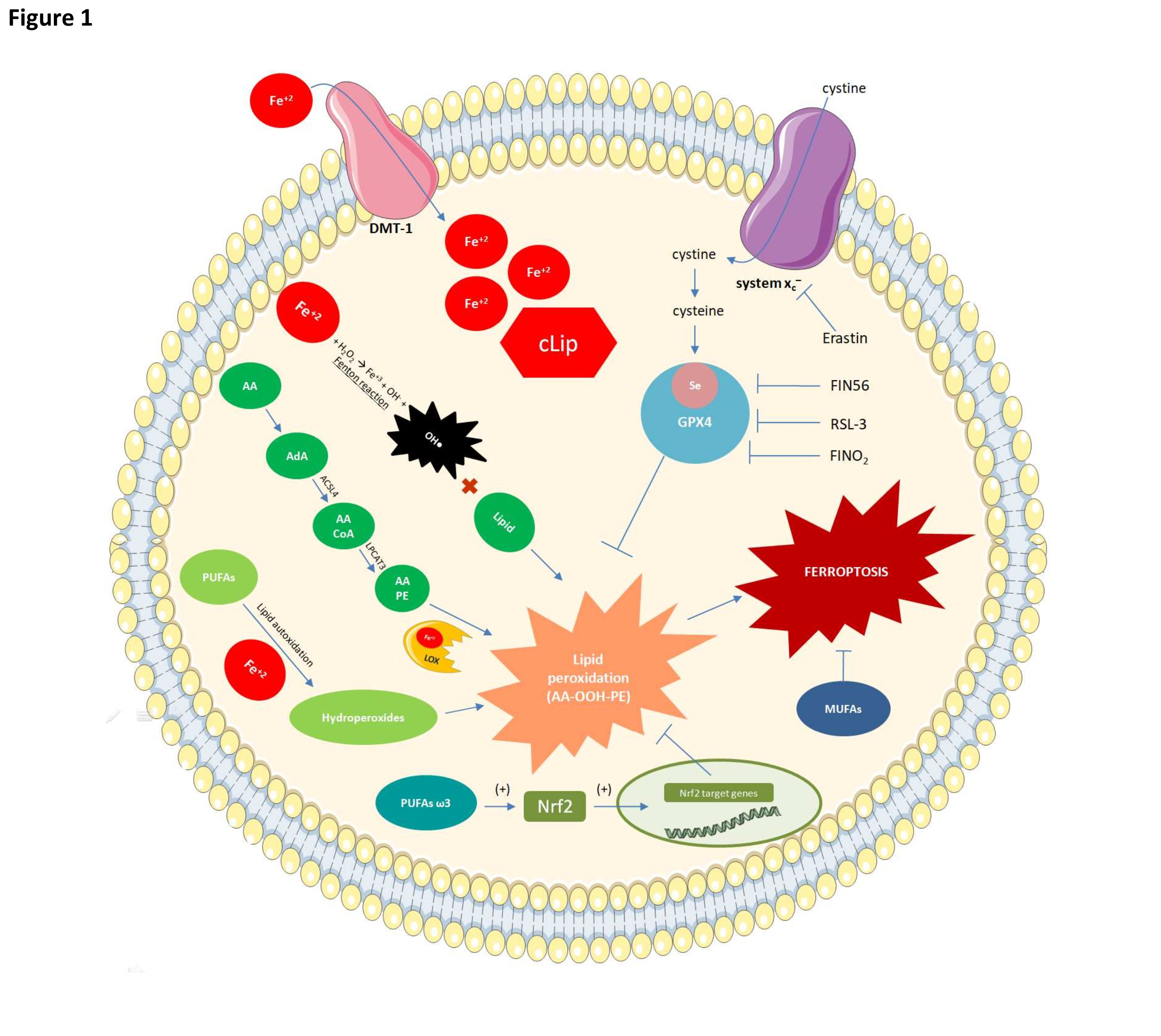
Fig. 1: Mechanisms of ferroptosis and their relationships with fatty acids. Glutathione peroxidase 4 (GPX4) inactivation renders cells susceptible to lipid peroxidation. Erastine may suppress GPX4, thereby preventing cystine from entering the cytosol. Cystine is required for glutathione synthesis and GPX4 activity. GPX4 may also be inhibited by RSL-3 which binds selenium (Se) at the active site of GPX4 and reduces its antioxidant activity. FIN56 degrades GPX4 while FINO2 indirectly inhibits GPX4 and oxidizes iron and lipids. Lipid peroxidation can occur by three distinct mechanisms of which the most common occurs via reactions involving Fe2+. The latter enters the cell via the DMT-1 protein. A cytoplasmic labile iron pool (cLIP) forms when there is excess intracellular iron. Fe2+ participates in the Fenton reaction to form hydroxyl radical which reacts with lipids to form lipid peroxides. In ferroptosis, elongases convert AA to AdA and the latter is oxidized by acyl-CoA synthase family 4 long chain (ACSL4) to arachidonoyl-CoA (AA-CoA). LPCAT3 then esterifies the latter to AA-PE. Fe-dependent lipoxygenases then oxidize the latter to the toxic phospholipid hydroperoxide AA-OOH-PE. Ferroptosis occurs when the AA-OOH-PE level exceeds the cell threshold. PUFAs also undergo auto-oxidation to lipid peroxides in the presence of iron. Omega-3 PUFAs induce the Nrf2 transcription factor (TF) which, in turn, promotes the synthesis of antioxidant enzymes such as GPX4. The latter detoxifies the phospholipid hydroperoxides responsible for ferroptosis. Monounsaturated fatty acids are relatively less susceptible to lipid peroxidation and their integration into membrane phospholipids can inhibit ferroptosis.
Iron metabolism
To determine the participation of ingested iron in ferroptosis the regulation of iron metabolism must first be clarified. Ingested iron is transformed into heme iron (Fe2+) and non-heme iron (Fe+3). The food sources of heme iron include red meats, offal (liver and kidney), poultry, pork, fish, and shellfish. Meat, dark green leafy vegetables and legumes are the main food sources of non-heme iron.When dietary non-heme iron (Fe+3) reaches the intestinal epithelium, it is reduced to the ferrous form (Fe2+) by cytochrome b duodenal ferrireductase (Dcytb) localized to the brush edges of duodenal enterocytes. The ferrous iron is then absorbed into the brush edges via the carrier protein divalent metal transporter 1 (DMT1) [16].The mechanism of intestinal heme absorption has not been fully elucidated. A putative heme transporter designated heme1 carrier protein (HCP1)was discovered in 2005. However, its role as an apical heme transporter was deemed unimportant as it also has a high affinity for folate and transports it [17]. Within the enterocytes, the heme iron is released from protoporphyrin via heme oxygenase. The released iron then is either stored as ferritin or is joined to the non-heme iron pool or released from the enterocytes into the blood. The protein ferroportin exports iron from the intestinal cells to the plasma. Ferroportin exports iron from enterocytes and macrophages [18, 19]. Hence, the Fe2+ out sourced by ferroportin is oxidized to Fe+3 by hephaestin and captured by transferrin [20].
Iron absorption in the enterocytes is regulated by negative feedback and involves hepcidin. This hormone is synthesized in the liver and maintains iron homeostasis by binding ferroportin [21]. The enterocytes internalize the hepcidin-ferroportin complex. Then, ferroportin degrades, iron efflux is reduced, and intestinal iron absorption and bioavailability decrease [21].
Senescent red blood cells (RBCs;erythrocytes) are also important iron sources. Most of the iron in the body is associated with hemoglobin. Thus, phagocytosis and degradation of senescent RBCs release iron [22]. Most of the iron in the human body is recovered from senescent erythrocytes through the splenic endoplasmic reticulum and the hepatic Kupffer cells [22]. Aging-related changes in erythrocytes include Band 3 membrane protein exposure, phosphatidylserine accumulation in the outer membrane, and membrane stiffness. All these are recognized by specific macrophage receptors [23]. Interaction of these receptors with RBCs causes the latter to undergo phagocytosis. Then, the RBC components are released. Heme accumulation in macrophage cytoplasms induces heme oxidase 1 which degrades heme into Fe2+, biliverdin, bilirubin, and carbon monoxide. Fe2+may be stored as ferritin in macrophages or exported by ferroportin [24]. In the latter case, the Fe2+ is oxidized to Fe+3 by the hepatic protein ceruloplasmin. The Fe+3 is then transported by transferrin mainly to the bone marrow where it participates in erythropoiesis [24].
Iron overload
Iron is an essential microelement with redox properties that are vital to several biological processes. Iron is a crucial component of hemoproteins such as myoglobin, hemoglobin, cytochrome p450, ironsulfur proteins and others involved in cellular metabolism.
There is no specific iron excretion mechanism under normal physiological conditions. Iron metabolism depends upon absorption from the diet (1-2 mg Fe/d) to compensate for Fe losses from menstrual bleeding and other blood loss [25]. Under aerobic conditions, iron that is not bound or sequestered by serum transferrin and cellular ferritin can freely interact with vascular, cellular, and subcellular structures and damage them. In iron overload, labile iron fractions with strong redox activity appear in the blood and cells. Labile iron in the plasma is not linked to transferrin. Intracellular iron is known as labile cell iron [26]. Iron that is not bound to transferrin may target hepatic and other parenchymatous tissue and is potentially cytotoxic [27]. Unbound iron is captured by parenchyma cells and especially hepatocytes, is not controlled by iron-regulating protein (IRP) or iron-responsive elements (IRE) [27], and accumulates in the cells. At transferrin saturation >75–80%, the iron is labile or in reactive plasma form [27].
Reactive plasma iron is a potentially toxic circulating form of the metal. Cytoplasmic iron induces ROS through the Fenton reaction and hydrogen peroxide is transformed into the highly reactive hydroxyl radical. Excess iron can damage cell membranes, mitochondria, nuclei, and DNA [28].
Certain hereditary or acquired pathological conditions disrupt cellular iron homeostasis and can lead to iron overload [26]. These disorders are associated with hemochromatosis which is the progressive and toxic accumulation of iron in parenchymatous organs. Primary iron overload results from a primary defect in iron homeostasis regulation. Secondary iron overload is associated with genetic factors and acquired diseases [29]. Hereditary hemochromatosis is characterized by increased intestinal iron absorption and progressive iron accumulation in various organs and tissues [30]. The most common mutation associated with hereditary hemochromatosis involves the gene regulating iron homeostasis. Other types of hereditary hemochromatosis involve mutations of the genes encoding hepcidin or ferroportin biosynthesis [30]. Secondary or acquired iron overload in hemoglobinopathies such as thalassemias and sickle cell disease is often characterized by anemia. Individuals with these conditions require regular blood transfusions [26]. However, this treatment may cause iron overload as each unit of transfused blood contains ~200–250 mg Fe [31].
Iron metabolism is altered in both primary hemochromatosis and secondary iron overload and may increase the morbidity and mortality of these conditions. Under iron overload, serum iron levels exceed the iron binding capacity of transferrin. Hence, the unbound iron content and the risk of cellular damage in the body increase. The main clinical manifestations of iron overload include liver and heart failure. Myocytes use Fe in myoglobin biosynthesis and are the second largest iron consumers in the body while hepatocytes intracellularly store up to 20% of all iron [25].
Iron supplementation
About 33% of the global population has anemia. Iron deficiency and inflammation are the main causes [32]. Iron deficiency is the leading cause of anemia and the most prevalent nutrient deficiency worldwide. It affects 33%,40%, and 42% of all non-pregnant women, pregnant women, and children, respectively [33]. In Brazil, anemia is prevalent in the 6–23 mo age group (19%) and affects ~ 17.3% of all pregnant women [34]. Iron deficiency occurs mainly as Fe requirements increase during periods of rapid growth and development such as early childhood, adolescence, and pregnancy. However, it may also occur at other stages of life [33]. Progressive iron deficiency can result in iron-deficient erythropoiesis, reductions in RBC counts and hemoglobin, fatigue, impaired physical performance, decreased productivity, and suboptimal brain development [33].
Inadequate dietary intake may result from an iron-poor diet and/or the ingestion of iron sources with low bioavailability. When iron deficiency anemia occurs, both dietary Fe sources and iron supplementation are necessary. Food may also be fortified with iron [35]. Iron-binding proteins such as transferrin and ferritin should accompany iron supplementation as excess unbound iron can be toxic. As there is no physiological mechanism for iron excretion, homeostatic iron regulation is mediated by the modulation of iron absorption mainly through the hepatic hormone hepcidin [32].
A randomized double-blind trial showed that 50 mg/d Fe supplementation for ~8 mo increased respiratory morbidity in South African children aged 6–11 y. However, the combination of iron supplementation, 420 mg/d DHA, and 80 mg/d EPA mitigated respiratory morbidity [36]. The same authors examined the biochemical effects of Fe and omega-3 polyunsaturated fatty acid (ω3-PUFA) supplementation both separately and in combination [37]. Omega-3 PUFA supplementation alone and in combination with Fe altered lipid-derived immunological modulator profiles, was anti-inflammatory, and reduced oxidative stress-related intermediate production [37]. By contrast, iron supplementation alone lowered serum anti-inflammatory mediator and prodrug concentrations [37]. Thus, ω3-PUFAs may confer protection against the oxidative damage caused by iron supplementation. However, the foregoing studies did not investigate the effects of Fe and ω3-PUFA supplementation on ferroptosis.
Intestinal absorption regulates ingested iron status. Hence, intestinal cells are the most susceptible to ferroptosis as the iron concentrations are well regulated in the body. Adults with normal bowel function are at low risk of developing dietary iron overload [38]. Nevertheless, high doses of iron supplements may have adverse effects on the gastrointestinal tract including abdominal discomfort, constipation, nausea, vomiting, or diarrhea [39]. Acute ingestion of >20 mg/kg Fe can cause corrosive intestinal necrosis, fluid and blood loss, shock, tissue damage, and organ failure [38]. The Food and Nutrition Board (FNB) of the Institute of Medicine (USA) established tolerable upper limits (ULs) for iron intake based on the negative gastrointestinal impact of excessive supplemental iron consumption. In pathological situations such as hemochromatosis that lead to primary iron overload, iron supplementation may cause ferroptosis in organs outside the gastrointestinal tract as well.
Effects of iron supplementation on intestinal physiology
Genetic and environmental factors can modulate the intestinal microbiota. Diet strongly influences gut microbial abundance and diversity [40]. The relationship between the intestinal microbiota and iron has been investigated [41]. The biodynamics and interaction between iron and the gut microbiota must be explored to clarify this relationship and develop novel iron deficiency treatments.
The World Health Organization (WHO) has recommended oral ferrous sulfate supplementation for children residing in areas with > 40% iron deficiency [42]. However, these recommendations have been re-evaluated as iron supplementation may increase the risks of infection, hospitalizations, and mortality [43-45]. A recent study compared various ferritin measurement methods [46]. For efficacious iron deficiency treatment by oral Fe administration, the effects of excess iron on the intestinal lumen and microbial structure and function must first be established [47]. Ferritin levels indicate iron reserves, deficiency, and overload and should be determined before initiating Fe supplementation to mitigate the risk and damage associated with iron overload.
Excess intestinal iron may occur in children undergoing prolonged Fe supplementation and especially in those with a genetic predisposition towards low iron uptake capacity [48]. Moreover, Fe supplementation has a substantial impact on the gut microbiota and may induce necrotizing enterocolitis (Fig. 2) [48]. Alterations in host iron homeostasis can affect the iron content in the intestinal lumen and the gut microbial composition. Changes in the latter could lead to infectious diseases [49]. Escherichia coli, Salmonella, Shigella are highly siderophilic bacterial pathogens [50]. By contrast, beneficial bacteria such as Bifidobacteria and Lactobacillus have low iron requirements [51] and create barriers against pathogen colonization in the gut [39]. Iron-rich environments increase the proliferation of other microbial pathogens (Fig. 2) [50]. Thus, low intraluminal iron content favors the growth of Lactobacillus and other beneficial bacteria while high intraluminal iron content promotes potentially pathogenic Bacteroides and E. coli (Fig. 2) [52]. Thus, excess unabsorbed iron in the colon lumen might have a negative impact on the host intestine-microbiota interface [39].
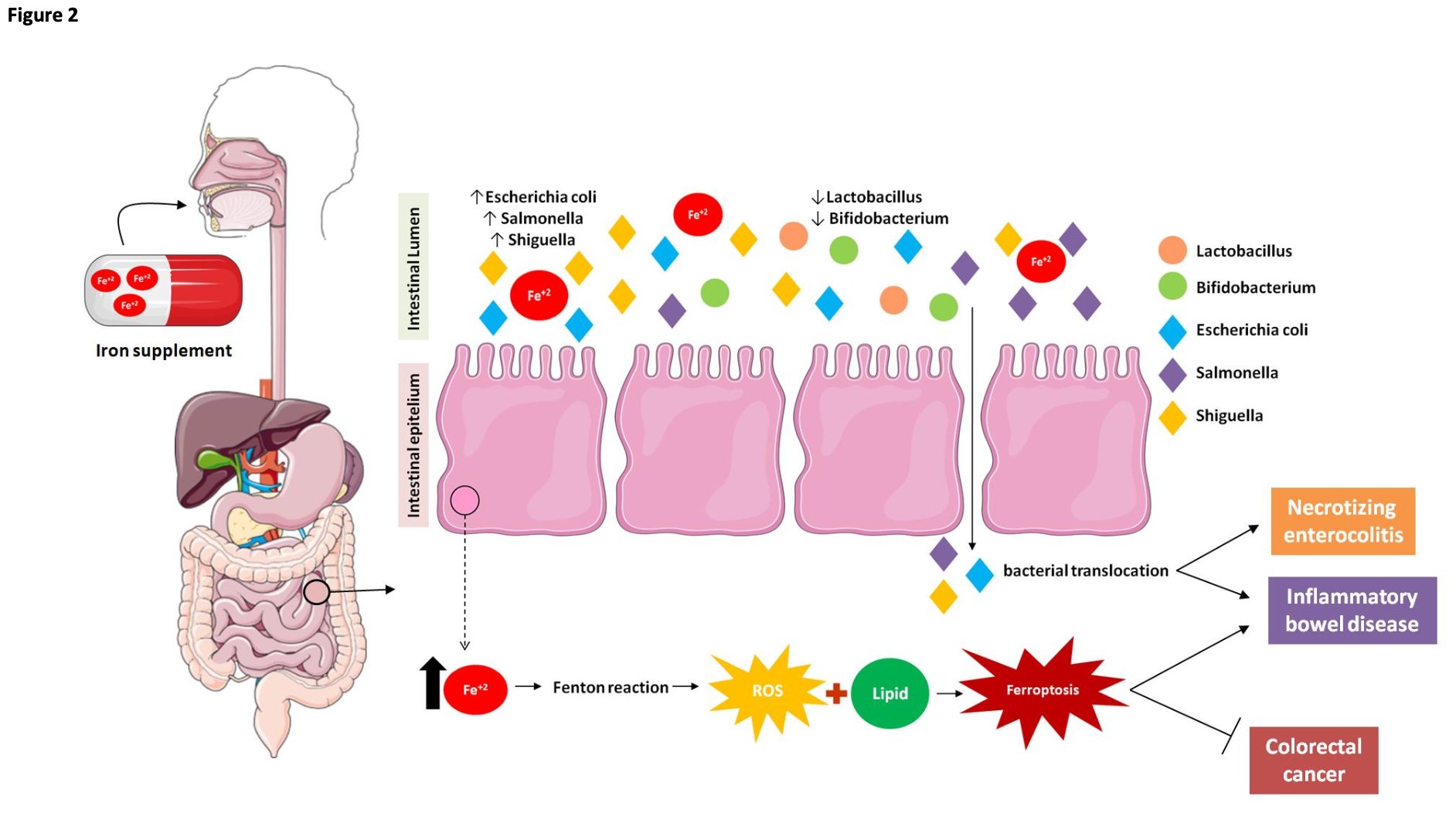
Fig. 2: Effect of oral iron supplementation on microbiota and intestinal cells. Alterations in host iron homeostasis can affect the iron content in the intestinal lumen and the gut microbial composition. Iron supplementation changes gut microbial homoeostasis and exacerbates siderophilic bacterial pathogens. Thus, these changes in the latter could lead to infectious diseases and may induce necrotizing enterocolitis. This alteration in the microbiota caused by excess intestinal iron is also associated with inflammatory bowel diseases. Mucosal ROS production is increased in inflammatory bowel disease (ulcerative colitis) in proportion to the disease activity, and iron chelators are known to reduce ROS production and ameliorate colonic symptoms in inflammatory bowel disease. Taken together, these findings suggest a possible relationship between inflammatory bowel disease and ferroptosis in which excess iron in the intestine produces ROS via the Fenton reaction, which triggers oxidative stress. Lipid peroxidation procedurally appears and ferroptotic cell death is induced. Thereby, the intestinal epithelial cells are destroyed, and damage to the intestinal mucosal barrier results in inflammatory bowel disease. However, it was observed that ferroptosis in a dose-dependent manner in different colorectal cancer cell lines promoted an increase in ROS and cellular labile iron pool (LIP) levels. Thus, ferroptosis allowed the inhibition of growth and spread of multiple types of cancer, such as colorectal cancer.
Other factors associated with luminal iron may also perturb the intestinal microbiota [47]. Fecal pH affects Fe solubility, reduction, and absorption [53]. Microbial galacto-oligosaccharide fermentation forms short-chain fatty acids (SCFAs), lowers pH, and facilitates Fe absorption in the gut [54]. SCFAs stimulate the proliferation of epithelial cells by increasing their absorptive surface. Prebiotics and their fermentation products upregulate iron-associated genes such as DMT1 in the duodenum and the colon [47]. As most of the non-heme dietary iron is not absorbed,it may be bioavailable to colonic bacteria [41].
Several studies have testified that excess oral irons can oxidize lipids [55]. Daily feeding 8000 µg of ferrous sulfate to normal rats increased malondialdehyde level by 1.6-fold [56]. Other studies have also authenticated that a daily iron supplement of 98 mg of iron in ferrous sulfate to women increased plasma malondialdehyde by more than 40% after 6 weeks of supplementation [57]. Furthermore, signs of gastrointestinal toxicity (nausea, vomiting, constipation, diarrhea, abdominal pain) have been observed following oral iron supplementation at different dosages in humans [55].
Excess dietary iron or oral iron supplementation may alter the intestinal phenotype and lead to inflammatory bowel diseases (IBD) mediated by iron and the regulation of ferroptosis [58]. On the other hand, iron supplementation is commonly administered in IBD and certain other intestinal pathologies to mitigate the anemia often associated with these conditions. However, studies in animals with a diet rich in iron (3, 000 mg/kg) identified that excess iron in the colonic lumen promoted increased inflammation and lipid peroxidation in colonic cells (Fig. 2) [59]. Taken together, these findings suggest a possible relationship between inflammatory bowel disease and ferroptosis in which excess iron in the intestine produces ROS via the Fenton reaction, which triggers oxidative stress (Fig. 2). Lipid peroxidation procedurally appears and ferroptotic cell death is induced.
Ferroptosis may positively or negatively regulate intestinal diseases depending on the cell type and the disease context [60]. Upregulation is associated with the fact that ferroptosis inhibits cancer growth [60]. It was observed that RSL3-induced ferroptosis in a dose-dependent manner in different colorectal cancer cell lines promoted an increase in ROS and cellular labile iron pool (LIP) levels [8]. Thus, ferroptosis allowed the inhibition of growth and spread of multiple types of cancer, such as colorectal câncer (Fig. 2).
Although iron supplementation has the theoretical potential to cause ferroptosis, the study by Horniblow et al. (2022) showed that iron supplementation could induce the expression of antioxidant genes colonocytes in vitro and the murine intestinal mucosa in mice fed with a high-iron diet [60]. Chronic iron exposure modified colonocyte epigenetics in vitro and the intestinal mucosae of mice fed an iron-rich diet (50 mg/kg ferrous sulfate) [60]. Evaluation of the methylation status of the CG locus disclosed significant changes in iron-dependent hypomethylation. The epigenetic changes at the CG locus were targeted by nuclear factor erythroid 2-related factor 2 (Nrf2) and activated NAD(P)H dehydrogenase quinone 1 (NQO1) and glutathione peroxidase 2 (GPX2). Hence, this mechanism might characterize the induction of the cellular antioxidant response to iron-mediated oxidative stress [60]. A qRT-PCR analysis of the main Nrf2 targets NQO1 and GPX2 revealed that their mRNA expression levels increased in populations administered iron [60]. Furthermore, the NQO1 and GPX2 protein expression levels were 9.4-fold and 2.8-fold higher, respectively, in Fe-treated than untreated populations [60].The findings were investigated in a murine model and in human intestinal tissue subjected to chronic high iron levels. Hence, excess iron from diet (50 mg/kg ferrous sulfate) and/or supplementation alters the intestinal phenotype and, by extension, modulates the regulation of ferroptosis [60].
Polyunsaturated fatty acids (PUFAs)
PUFA metabolism
Fatty acids are hydrocarbons varying in chain length, double bond number and position, and degree of saturation. Mammals cannot synthesize LA and ALA from the precursor oleic acid and the conversion efficiency of LA and ALA to AA, DHA, and EPA is low. Therefore, direct uptake from diet appears to be significantly more effective [61]. They are classified based on the positions of the double bonds in their carbon chains and are designated omega-3 (ω3) and omega-6 (ω6) PUFAs [61]. PUFAs have ≥ 2 double bonds and are structural components of membrane phospholipids [61]. PUFAs enable the flexibility, fluidity, permeability, and modulation of the proteins embedded in cell and organellar membranes. They also increase the number of receptors and their affinity for the hormones and growth factors they bind [62]. The organization of biological membranes depends on the length and degree of unsaturation of their embedded fatty acid chains. PUFAs are highly disordered compared to saturated and monounsaturated fatty acids. Thus, their presence in membrane phospholipids enhances membrane flexibility [63].
PUFAs are also precursors of lipid mediators of inflammation, vascular permeability, chemotaxis, and vasoconstriction [62]. Omega-3 PUFAs are long-chain hydrocarbons with carboxylic acids at one end (alpha terminal) and methyl groups at the other (omega terminal). They have ≥ 2 double bonds in the third position (ω-3 or n-3) [64]. Desaturases convert omega-3 PUFA ALA into EPA and the latter into DHA. However, these reactions are slow and inefficient [64]. Therefore, dietary sources of ALA are beneficial and include flaxseed, chia, nuts, and their oils [65]. An adequate amount of ALA is equivalent to 0.5% of the total caloric intake from lipids and should prevent the development of deficiency symptoms [66]. The consumption of fatty marine fish such as mackerel, salmon, and tuna as well as fish oil supplies EPA and DHA [65].
Omega-3 PUFAs are implicated in several mechanisms involving the cell membrane, the cytosol, and the nucleus [67] since they bind receptors and regulate signaling processes influencing gene expression [67]. Alterations in cell membrane fatty acid composition affect RBC aggregation and deformability, membrane elasticity, and the inflammatory response [67, 68].
Omega-6 PUFAs include linoleic acid which occurs in soybean and sunflower seeds and other plant foods. An adequate amount of linoleic acid is equivalent to 2–3% of the total caloric intake from lipids and should prevent the development of deficiency symptoms [66]. The total intake of omega-6 PUFAs should constitute 2.5–9% of the total caloric value from lipids [66]. Linoleic acid per se has both physiological action and health benefits.It is also a substrate for the biosynthesis of other omega-6 PUFAs such as γ-linolenic acid (18:3ω-6), dihomo-γ-linolenic acid (20:3ω-6) and AA [69]. The latter also occurs in meat and eggs. Omega-6 PUFAs may be incorporated into membrane phospholipids. However, AA is typically present in relatively larger amounts there [61]. Arachidonic acid reacts with molecular oxygen via the cyclooxygenase, lipoxygenase, and cytochrome P450 pathways and generates eicosanoids including prostaglandins, thromboxanes, and leukotrienes. The aforementioned substances induce inflammation, vasoconstriction, and platelet aggregation [69].
Purposes of fatty acids supplementation
In a typical Western diet, the intake of linoleic acid is five-fold to 15-fold higher than that of ALA. Consequently, this lifestyle favors proinflammatory mediator production, vasoconstriction, and platelet aggregation [69]. Hence, the proportion of omega-6:omega-3 PUFAs is 15:1 in the West [61]. A prior study demonstrated that morbidity and mortality decrease with omega-6:omega-3 ratio [7]. The most recent WHO report stated that there is limited empirical evidence for the benefit of applying the omega-6:omega-3 ratio or recommending a specific dietary LA:ALA ratio [66]. The report also indicated that intake of the PUFAs within the established quantities suffices to maintain good health [66]. Excessive omega-6 PUFAs and very high omega-6:omega-3 ratios promote cardiovascular diseases (CVDs), cancers, and inflammatory and autoimmune diseases [7]. By contrast, diets with adequate omega-3 PUFAs and low omega-6:omega-3 ratios are anti-inflammatory and lower the risks of developing the foregoing diseases [61].
Supplements containing omega-3 PUFAs have been prescribed to manage diseases associated with inflammation. Omega-3 PUFAs have been supplied as EPA or DHA or in the form of fish oils. An increase in the incorporation of omega-3 PUFAs into cell membranes is associated with decreased prostaglandin and leukotriene production and modulation of the inflammatory process [68]. EPA and DHA also inhibit NFκB activation [68]. They activate the anti-inflammatory transcription factor peroxisome proliferator-activated receptor gamma (PPAR-γ). They disrupt sphingolipid- and cholesterol-rich regions and microdomains in the plasma membrane that initiate inflammatory signaling and reduce the fluidity of the membrane by increasing its saturated FA and cholesterol content [68]. EPA and DHA also activate the gene encoding the G-protein coupled receptor 120 (GPR120) which is expressed in inflammatory macrophages and in vivo when obese mice are supplemented with these PUFAs (isocaloric high fat diet containing 27% fish oil supplementation enriched with 50 and 100 mg of DHA and EPA, respectively, per mouse, per Day) [70]. Hence, EPA and DHA are involved in the inflammatory cascade mediated by GRP120. In this manner, they block the signaling pathway that activates NK-κB [68, 70]. NF-κB inhibition is related to a reduction in proinflammatory cytokine biosynthesis, a decrease in adhering molecule expression, downregulation of the gene encoding cyclooxygenase-2, and the induction of nitric oxide (NO) synthetase [68]. EPA and DHA are also antioxidants. They stabilized RNAm coding for Nrf2 when a total of 2, 700 mg/day of omega-3 PUFAswas supplemented in subjects with type 2 diabetes [71]. Nrf2 transcribes the genes encoding cytoprotective proteins such as catalase, glucose 6-phosphate dehydrogenase, superoxide dismutase, thioredoxin, glutathione peroxidase, and glutathione. For these reasons, omega-3 PUFAs supplementation has been widely tested and used.
Effects of omega-3 PUFA supplementation on ferroptosis
Omega-3 PUFA supplementation could be effective considering the health benefits of including these substances in the diet and the fact that their intake levels are low in the West. However, concern has been raised regarding EPA and DHA supplementation when there is excess iron in the body [72] because lipid peroxides are derived from the oxidation of the PUFAs in cell membranes [6]. In addition, the accumulation of lipid peroxidation by-products increases the porosity, destroys the barrier function, thins, and alters the permeability of membranes [73].
In tumor models, certain lipid-based mechanisms cooperated to increase or decrease the susceptibility to ferroptosis [6]. Increased lipid absorption can enrich the PUFA content of cancer cell membranes, thereby promoting ferroptosis [6]. Long-chain-fatty-acid—CoA ligase 4 (ACSL4) incorporates PUFAs into lipid membranes and increases the susceptibility of tumor cells to ferroptosis [6]. Other mechanisms such as System Xc- inhibition, blockage of cellular cystine penetration, and reduction of glutathione synthesis are associated with alterations in TFs. These events augment the susceptibility of tumor cells to ferroptosis [74]. However, certain cancer models are resistant to ferroptosis [75] and are characterized by an aggressive tumor type. Ferroptosis resistance in tumor cells is related to changes in lipid metabolism and augmented lipogenesis generating saturated and monounsaturated fatty acids. These Acyl-CoA Synthetase Long Chain Family Member 3 (ACSL3)-mediated modifications enrich cancer cell membranes with phospholipids and induce ferroptosis resistance (Fig. 1) [6, 76]. Ferroptosis resistance is also associated with high antioxidant potential created by cystine transport, glutathione biosynthesis, and GPX4 in tumor cells (Fig. 1) [6, 16].
There is no published evidence for prior clinical trials evaluating the effects of omega-3 PUFA supplementation on ferroptosis. However, various tumors subjected to omega-3 PUFAs presented with increased lipogenic enzyme expression and activity that could be associated with tumor development in experimental studies [6].
The degree of unsaturation of cell membrane FAs may determine whether tumor cells proliferate or die [6]. Membrane phospholipid composition can influence cell surface receptor signaling such as epidermal growth factor receptor (EGFR) grouping [6]. Saturated FAs are associated with tumor development [77] whereas long-chain PUFAs such as AA and EPA were cytotoxic to glioblastomas and other tumors [78]. Hence, membrane structure and sphingolipid and cholesterol content, phospholipid FA profiles, and lipogenic enzymes strongly influence oncogenic signaling and, therefore, tumor development. Omega-3 PUFAs can stabilize Nrf2 and reduce lipid peroxidation (Fig. 1) [71, 79]. However, the same PUFAs may themselves be susceptible to lipid peroxidation. EPA and DHA form micelles that eliminate free radicals and diminish hydroxyl and superoxide radical production [80]. Evidence from in vivo and in vitro studies demonstrates that EPA and DHA downregulate NADPH oxidase which contributes to oxidative stress [80]. Though these unsaturated FAs are susceptible to peroxidation, they can nonetheless reduce free radicals, oxidative stress, and ferroptosis. EPA and DHA supplementation did not increase lipid peroxides (lipid hydroperoxides, malondialdehyde (MDA), and isoprostanes) in patients with coronary artery disease, when they received 1.9g/day omega-3 PUFAs [81]. A randomized double-blind trial showed that EPA and DHA intervention (6g/d omega-3 PUFAs) increased EPA- and DHA-rich phospholipids and lowered arachidonic and linoleic acid levels [82]. Therefore, omega-3 PUFA supplementation may lower the incidence of ferroptosis as it lowers the concentration of AA. The latter is the main FA implicated in ferroptosis. The same study reported that 6 wks of EPA and DHA supplementation lowered the MDA and lipid peroxide levels. This finding upheld the aforementioned hypotheses [82]. Glutathione peroxidase activity did not change after this intervention [82].
A non-insulin-dependent diabetic population was administered 1, 080 mg/d EPA and 720 mg/d DHA. The patients presented with reductions in MDA concentration and increases in glutathione peroxidase but no alteration in RBC catalase or superoxide dismutase (SOD) activity [83]. Thus, omega-3 PUFAs increase the antioxidant potential and reduce lipid peroxidation [83].
A randomized, placebo-controlled, double-blind trial on type 2 diabetic patients administered 600 mg/d EPA and 300 mg/d DHA supplementation for 10 wks disclosed that their Nrf2 expression levels were higher than those of placebo-treated controls [71]. The diabetic patients also displayed relative decreases in MDA level and increases in total antioxidant capacity. Hence, the omega-3 PUFA treatment reduced oxidative stress [71]. Fish oil supplementation had similar effects on patients with breast cancer [84]. The foregoing findings demonstrate a possible relationship between omega-3 PUFAs and the reduction of ferroptosis. Moreover, Nrf2 inhibited ferroptosis in cancer cells (Fig. 1) [85]. Another study investigated the relationship between monounsaturated fatty acids and ferroptosis [76]. In human HT-1080 cells, cell death induced by the pro-ferroptotic System Xc- inhibitor erastin was suppressed by cotreatment with the MUFAs oleic acid (18:1 cis-9) and palmitoleic acid (16:1 cis-7)(Fig. 1) [76]. Exogenous oleic acid suppressed ferroptosis induced by a more potent erastin analog in other cell lines such as T98G glioblastoma and A549 non-small cell lung carcinoma cells, as well as in fibroblasts. Oleic acid blocks the GPX4 inhibitor erastin and competes against PUFAs for incorporation into phospholipids. ACSL3 integrates exogenous monounsaturated fatty acids into phospholipids, thereby reducing PUFA incorporation into phospholipids, displacing them from cell membranes, preventing lipid accumulation, and inhibiting ferroptosis. Exogenous monounsaturated fatty acids protect cells against apoptotic lipotoxicity caused by ACSL3-independent saturated FA accumulation [86]. Monounsaturated FAs are not substrates for the lipid peroxidation that occurs during ferroptosis [76].
Interaction between iron metabolism and PUFAs
The liver metabolizes iron and omega-3 PUFAs. It synthesizes and activates enzymes implicated in Fe and PUFA metabolism. Interactions between PUFAs and Fe occur in response to supplementation with iron and/or omega-3 PUFAs [87, 88].
Omega-3 and omega-6 PUFAs decreased in most tissues and especially the RBCs of animals fed an iron-rich diet (200 mg Fe/kg food/d). The exception was a decrease in the long-chain PUFA content of adipose tissue [89]. The same study reported that the brain had the highest resistance to alterations in FA profile [89]. EPA and DHA were dramatically reduced in all tissues and particularly the RBCs and adipose tissue [89]. High Fe doses downregulated the Δ5 and Δ6 desaturases required for EPA and DHA synthesis, respectively (Fig. 3A). This mechanism may explain the observed reductions in endogenous FA synthesis in response to high iron dosage (Fig. 3A) [89]. These desaturases normally have low activity (Fig. 3A). Nevertheless, iron-rich diets increased their mRNA expression levels compared to the control diet (50 mg Fe/kg food/d) [87]. Free radicals might have inactivated these desaturases. Desaturase activity was negatively correlated with oxidative stress and positively correlated with glutathione concentration. Desaturase inactivation might have decreased endogenous EPA and DHA biosynthesis. Increases in desaturase mRNA expression may indicate adaptive responses to diminished enzyme activity. The findings of Valenzuela et al. (2018) corroborated the ferroptosis mechanism. Excess iron increases oxidative stress and leads to PUFA oxidation (Fig. 3A).
In rodents, iron supplementation (200mg Fe/kg food/d) lowered the omega-6 and omega-3 PUFA concentrations, raised the omega-6:omega3 ratios, and decreased hepatic and adjacent extrahepatic FA unsaturation and Δ5 and Δ6 desaturase activity [90]. By contrast, extra virgin olive oil supplementation (100 mg/d) prevented increases in the mRNA expression levels of both desaturases in rodents who received an iron-rich diet [90]. Thus, supplementation with antioxidant FAs may prevent the oxidative changes induced by Fe-rich diets.
Changes in intracellular iron content altered the expression levels of certain genes regulating lipid metabolism (Fig. 3A). In certain animals, iron-rich diet increased the activity of the sterol regulatory element SREBP-1 (Fig. 3A). Consequently, endogenous FA biosynthesis was increased [87], such as palmitic acid (C16:0); palmitoleic acid (C16:1n7); oleic acid (C18:1n9); vaccenic acid (C18:1n7) [91]. SREBP-1 promotes the transcription and increases the plasma level of hepcidin which binds ferroportin. The latter then degrades and iron accumulates in the liver (Fig. 3A) [92]. Omega-3 PUFAs have the opposite effect (Fig. 3B). They decrease the SREBP, hepcidin, and intracellular iron levels (Fig. 3B)[92] . However, the mechanism of this phenomenon is unknown. On the other hand, an inverse association was observed between serum concentrations of omega-3 PUFAs and serum interleukin-6 (IL-6) concentrations in an observational study [93] and, by extension, concentrations of omega 3 PUFAS decrease hepcidin expression [94]. Omega-3 PUFA supplementation may reduce intracellular Fe by decreasing the susceptibility to ferroptosis (Fig. 3B).
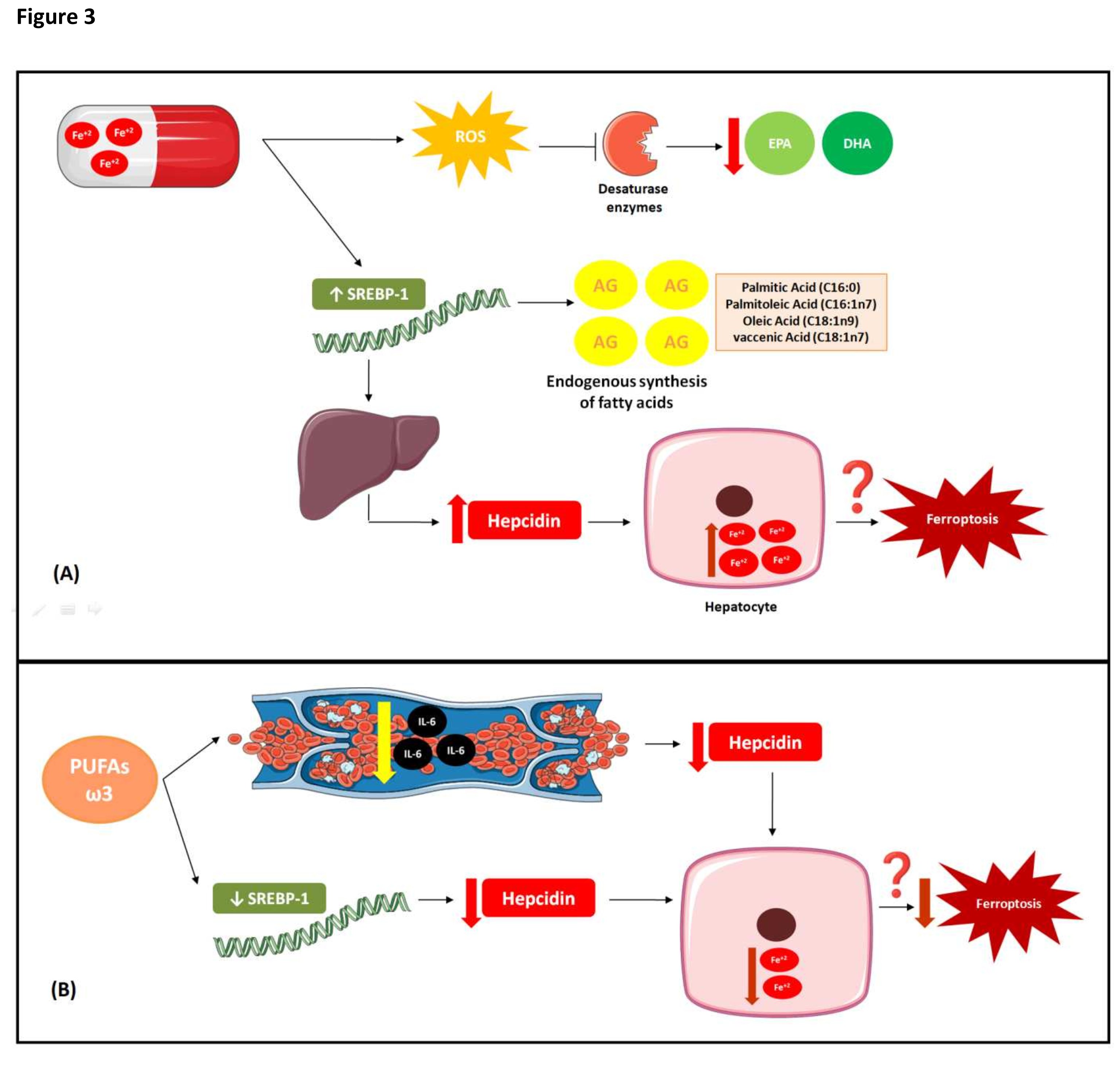
Fig. 3: Interaction between iron and PUFAs metabolisms. (A) Interaction between iron metabolism and PUFAs under conditions of high Fe doses High doses of Fe in animal studies decreased the desaturases required for eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA) and synthesis, which may explain the reductions in endogenous FA synthesis in response to high iron dosage. The free radicals produced can inactivate these desaturases. Changes in intracellular iron content altered the expression levels of certain genes regulating lipid metabolism. Iron-rich diet increased the activity of the sterol regulatory element-binding transcription factor 1(SREBP-1), which promoted an increase in endogenous FA biosynthesis (palmitic acid; palmitoleic acid; oleic acid; vaccenic acid). SREBP-1 promotes the transcription and increases the plasma level of hepcidin which binds ferroportin. The latter then degrades and iron accumulates in the liver, which can lead to increased susceptibility to ferroptosis.(B) Effects of Omega-3 PUFA on iron metabolism. Omega-3 PUFAs have the opposite effect of high Fe dosages. They decrease the SREBP, hepcidin, and intracellular iron levels. Although the mechanism of this phenomenon is unknown, omega-3 PUFAs might lower serum interleukin-6 (IL-6) concentrations and, by extension, decrease hepcidin expression. Thus, Omega-3 PUFA supplementation may reduce intracellular Fe by decreasing the susceptibility to ferroptosis.
Conclusion
PUFAs are protagonists in lipid peroxidation and ferroptosis. Nevertheless, omega-3 PUFAs do not apparently contribute to these processes even in the presence of excess iron. Omega-3 PUFAs activate transcription factors associated with the upregulation of antioxidant enzymes. On the other hand, excess iron supplementation is usually toxic to the intestinal epithelium and microbiota. Few studies have investigated the impact of diet and supplementation on ferroptosis. Controlling ferroptosis determines whether cells proliferate or die. Therefore, future studies should endeavor to establish the roles of supplementation and diet in its occurrence.
Acknowledgements
The authors thank the excellent technical assistance provided by Dr. Joana Pereira and MSc. Viviane Fernandes.
Author Contributions
ED, JDCM, SMLF, ICP and MSB were responsible for conducting the search, screening potentially eligible studies, extracting and analyzing data, interpreting results. ED wrote the first draft of the manuscript. ED and MC were responsible for drafting the manuscript, analyzing data and interpreting results.
Funding Sources
This study was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (Marta Citelli, process# 408401/2017-6); Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) (Marta Citelli -process # E-26-211.844/2021).
Disclosure Statement
No potential Disclosure Statement was reported by the authors.
References
| 1 | Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, Stockwell BR: Ferroptosis: an iron-dependent form of non apoptotic cell death. Cell 2012;149:1060-72.
https://doi.org/10.1016/j.cell.2012.03.042 |
| 2 | Green DR: An element of life. Cell 2018;172:389-390.
https://doi.org/10.1016/j.cell.2018.01.003 |
| 3 | Lei P, Bai T, Sun Y: Mechanisms of ferroptosis and relations with regulated cell death: a review. Front Physiol 2019;10:139.
https://doi.org/10.3389/fphys.2019.00139 |
| 4 | Muckenthaler MU, Rivella S, Hentze MW, Galy B: A Red Carpet for Iron Metabolism. Cell 2017;168:344-361.
https://doi.org/10.1016/j.cell.2016.12.034 |
| 5 | Pietrangelo, A: Hereditary Hemochromatosis: Pathogenesis, Diagnosis, and Treatment. Gastroenterology 2010;393-408.
https://doi.org/10.1053/j.gastro.2010.06.013 |
| 6 | Broadfield LA, Pane AA, Talebi A, Swinnen JV, Fendt SM: Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev Cell 2021;56:1363-1393.
https://doi.org/10.1016/j.devcel.2021.04.013 |
| 7 | Simopoulos, AP: The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Biomed Pharmacother 2002;56:365-379.
https://doi.org/10.1016/S0753-3322(02)00253-6 |
| 8 | Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M, Han X, Xiang Y, Huang X, Lin H, Xie T: RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front Pharmacol 2018.
https://doi.org/10.3389/fphar.2018.01371 |
| 9 | Li J, Cao F, Yin H, Huang Z, Mao N, Sun B, Wang G: Ferroptosis: past, present and future. Cell Death Dis 2020;11:1-13.
https://doi.org/10.1038/s41419-020-2298-2 |
| 10 | Jia M, Qin D, Zhao C, Chai L, Yu Z, Wang W, Tong L, Lv L, Wang Y, Rehwinkel J, Yu J, Zhao W: Redox homeostasis maintained by GPX4 facilitates STING activation. Nat Immunol 2020;21:727-735.
https://doi.org/10.1038/s41590-020-0699-0 |
| 11 | Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang J, Anthonymuthu T, Mohammadyani D, Yang Q,Proneth B, Klein-Seetharaman J, Watkins S, Bahar I, Greenberger J, Mallampalli RK, Stockwell BR, Tyurina YY, Conrad M, Bayır H: Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 2017;13:81-90.
https://doi.org/10.1038/nchembio.2238 |
| 12 | Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, Stockwell BR: Human haploid cell genetics reveals roles for lipid metabolism genes in non apoptotic cell death. ACS Chem Biol 2015;10:1604-1609.
https://doi.org/10.1021/acschembio.5b00245 |
| 13 | Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, Heindel DW, Zuckerman DS, Bos PH, Reznik E, Ye LF, Tyurina YY, Lin AJ, Shchepinov MS, Chan AY, Peguero-Pereira E, Fomich MA, Daniels JD, Bekish AV, Shmanai VV, Kagan VE, Mahal LK, Woerpel KA, Stockwell BR: FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol 2018;14:507-515.
https://doi.org/10.1038/s41589-018-0031-6 |
| 14 | Soupene E, Kuypers FA: Mammalian long-chain acyl-CoA synthetases. Exp Biol Med 2008;233:507-521.
https://doi.org/10.3181/0710-MR-287 |
| 15 | Shindou H, Shimizu T: Acyl-CoA: lysophospholipid acyltransferases. J Biol Chem 2009; 284:1-5.
https://doi.org/10.1074/jbc.R800046200 |
| 16 | Fleming MD, Trenor CC, Su MA, Foernzler D, Beier DR, Dietrich WF, Andrews NC: Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet 1997;16:383-386.
https://doi.org/10.1038/ng0897-383 |
| 17 | Blanc SL, Garrick MD, Arredondo M: Heme Carrier protein 1 transports heme and is involved in heme-Fe metabolism. Am J Physiol Cell Physiol 2012;302:1780-1785.
https://doi.org/10.1152/ajpcell.00080.2012 |
| 18 | Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BW, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI: Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000;403:776-781.
https://doi.org/10.1038/35001596 |
| 19 | McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, Hediger MA, Hentze MW, Simpson RJ: A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 2000;5:299-309.
https://doi.org/10.1016/S1097-2765(00)80425-6 |
| 20 | Fuqua BK, Lu Y, Darshan D, Frazer DM, Wilkins SJ, Wolkow N, Bell AG, Hsu JA, Yu CC, Chen H, Dunaief JL, Anderson GJ, Vulpe CD: The multi copper ferroxidase hephaestin enhances intestinal iron absorption in mice. PLoS ONE 2014; 9:1-13.
https://doi.org/10.1371/journal.pone.0098792 |
| 21 | Camaschella C, Nai A, Silvestri L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020;105:260-272.
https://doi.org/10.3324/haematol.2019.232124 |
| 22 | Mertens C, Marques O, Horvat NK, Simonetti M, Muckenthaler MU, Jung M: The Macrophage Iron Signature in Health and Disease. Int J Mol Sci 2021;22:1-30.
https://doi.org/10.3390/ijms22168457 |
| 23 | Foller M, Huber SM, Lang F: Erythrocyte programmed cell death. IUBMB Life 2008;60:661-668.
https://doi.org/10.1002/iub.106 |
| 24 | Winn NC, Volk KM, Hasty AH: Regulation of Tissue Iron Homeostasis: The Macrophage "Ferrostat". JCI Insight 2020;5:1-14.
https://doi.org/10.1172/jci.insight.132964 |
| 25 | Mancardi D, Mezzanotte M, Arrigo E, Barinotti A, Roetto A: Iron overload, oxidative stress, and ferroptosis in the failing heart and liver. Antioxidants 2021;10:1-18.
https://doi.org/10.3390/antiox10121864 |
| 26 | Corradini E, Buzzetti E, Pietrangelo A: Genetic iron overload disorders. Mol Aspects Med 2020;75:1-10.
https://doi.org/10.1016/j.mam.2020.100896 |
| 27 | Brissot P, Troadec MB, Loréal O, Brissot E: Pathophysiology and classification of iron overload diseases; update 2018. Transfus Clin Biol 2019;26:80-88.
https://doi.org/10.1016/j.tracli.2018.08.006 |
| 28 | Troadec MB, Loreal O, Brissot P: The interaction of iron and the genome: for better and for worse. Mutat Res Ver Mutat Res 2017;774:25-32.
https://doi.org/10.1016/j.mrrev.2017.09.002 |
| 29 | Piperno A: Classification and diagnosis of iron overload. Haematolgica 1998;83:447-455. |
| 30 | Fowler C: Hereditary hemochromatosis: pathophysiology, diagnosis, and management. Crit Care Nurs Clin North Am 2008;20:191-201.
https://doi.org/10.1016/j.ccell.2008.01.003 |
| 31 | Remacha A, Sanz C, Contreras E, De Heredia CD, Grifols JR, Lozano M, Nuñez GM, Salinas R, Corral M, Villegas A: Guidelines on haemovigilance of post-transfusional iron overload. Blood Transfus 2013;11:128-139. |
| 32 | Ganz T: Systemic iron homeostasis. Physiol Ver 2013;93:1721-1741.
https://doi.org/10.1152/physrev.00008.2013 |
| 33 | World Health Organization Internet: WHO guideline on use of ferritin concentrations to assess iron status in individuals and populations. World Health Organization 2020.
URL: https://www.who.int/publications/i/item/9789240000124 |
| 34 | Federal University of Rio de Janeiro Internet. Biomarkers of micronutrient status: prevalence of deficiencies and micronutrient distribution curves in Brazilian children under 5 years of age: ENANI 2019. Federal University of Rio de Janeiro 2021.
URL: https://enani.nutricao.ufrj.br/index.php/relatorios/ |
| 35 | World Health Organization and Food and Agriculture Organization of the United Nations Internet. Guidelines on food fortification with micronutrients. World Health Organization 2006.
URL: https://www.who.int/publications/i/item/9241594012 |
| 36 | Malan L, Baumgartner J, Calder PC, Zimmermann MB, Smuts CM: n-3 Long-chain PUFAs reduce respiratory morbidity caused by iron supplementation in iron-deficient South African schoolchildren: A randomized, double-blind, placebo controlled intervention. Am J Clin Nutr 2015;101:668-679.
https://doi.org/10.3945/ajcn.113.081208 |
| 37 | Malan L, Baumgartner J, Zandberg L, Calder PC, Smuts CM: Iron and a mixture of DHA and EPA supplementation, alone and in combination, affect bioactive lipid signalling and morbidity of iron deficient South African schoolchildren in a two-by-two randomized controlled trial. Prostaglandins Leukot Essent Fatty Acids 2016;105:15-25.
https://doi.org/10.1016/j.plefa.2015.12.005 |
| 38 | Aggett PJ. Iron, in: Erdman JW, Macdonald IA, Zeisel SH (eds): Present Knowledge in Nutrition. 10th ed. Wiley-Blackwell, 2012, pp 506-520.
https://doi.org/10.1002/9781119946045.ch33 |
| 39 | Institute of Medicine Internet. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc: a Report of the Panel onMicronutrients. The National Academies Press. 2001.
URL: https://nap.nationalacademies.org/catalog/10026/dietary-reference-intakes-for-vitamin-a-vitamin-k-arsenic-boron-chromium-copper-iodine-iron-manganese-molybdenum-nickel-silicon-vanadium-and-zinc |
| 40 | Power SE, O'Toole PW, Stanton C, Ross RP, Fitzgerald GF: Intestinal microbiota, diet and health. Br J Nutr 2014;111:387-402.
https://doi.org/10.1017/S0007114513002560 |
| 41 | Puga AM, Samaniego-Vaesken ML, Montero-Bravo A, Ruperto M, Partearroyo T, Varela-Moreiras G: Iron Supplementation at the Cross roads of Nutrition and Gut Microbiota: The State of the Art. Nutrients 2022;14:1926.
https://doi.org/10.3390/nu14091926 |
| 42 | World Health Organization Internet: Guideline: Daily iron supplementation in infants and children. World Health Organization 2016.
URL: https://www.who.int/publications/i/item/9789241549523 |
| 43 | World Health Organization Internet: Guideline: Intermittent iron and folic acid supplementation in non-anemic pregnant women. World Health Organization 2013.
URL: https://apps.who.int/iris/bitstream/handle/10665/75335/9789248502019_por.pdf;jsessionid=420ABE0939AF95BFC92302218CDE7850?sequence=9 |
| 44 | Gera T, Sachdev HPS: Effect of iron supplementation on incidence of infectious illness in children: Systematic review. BMJ 2002;325:1142.
https://doi.org/10.1136/bmj.325.7373.1142 |
| 45 | Iannotti LL, Tielsch JM, Black MM; Black RE: Iron supplementation in early childhood: Health benefits and risks. Am J Clin Nutr 2006;84:1261-1276.
https://doi.org/10.1093/ajcn/84.6.1261 |
| 46 | World Health Organization: WHO guidance helps detect iron deficiency and protect brain development. World Health Organization 2020.
URL: https://www.who.int/news/item/20-04-2020-who-guidance-helps-detect-iron-deficiency-and-protect-brain-development |
| 47 | Kortman GA, Raffatellu M, Swinkels DW, Tjalsma H: Nutritional iron turned inside out: Intestinal stress from a gut microbial perspective. FEMS Microbiol Ver 2014;38:1202-1234.
https://doi.org/10.1111/1574-6976.12086 |
| 48 | Göpel W, Drese J, Rausch TK, Twisselmann N, Bohnhorst B, Müller A, Franz A, Ziegler A, Härtel C, Herting E: Necrotizing enterocolitis and high intestinal iron uptake due to genetic variants. Pediatr Res 2018;83:57-62.
https://doi.org/10.1038/pr.2017.195 |
| 49 | Buhnik-Rosenblau K, Moshe-Belizowski S, Danin-Poleg Y, Meyron-Holtz EG: Genetic modification of iron metabolism in mice affects the gut microbiota. Bio Metals 2012;25:883-892.
https://doi.org/10.1007/s10534-012-9555-5 |
| 50 | Andrews SC, Robinson AK, Rodriguez-Quinones F: Bacterial iron homeostasis. FEMS Microbiol Rev 2003;27:215-237.
https://doi.org/10.1016/S0168-6445(03)00055-X |
| 51 | Bezkorovainy A, Solberg L: Ferrous iron uptake by Bifidobacterium breve. Biol Trace Elem Res 1989;20:251-67.
https://doi.org/10.1007/BF02917440 |
| 52 | Paganini D, Zimmermann MB.: The effects of iron fortification and supplementation on the gut microbiome and diarrhea in infants and children: A review. Am J Clin Nutr 2017;106:1688S-1693S.
https://doi.org/10.3945/ajcn.117.156067 |
| 53 | Salovaara S, Sandberg AS, Andlid T: Combined impact of pH and organic acids on iron uptake by Caco-2 cells. J Agric Food Chem 2003;51:7820-7824.
https://doi.org/10.1021/jf030177n |
| 54 | Perez-Conesa D, Lopez G, Ros G: Effect of probiotic, prebiotic and synbiotic follow-up infant formulas on iron bioavailability in rats. Food Sci Technol Int 2007;13:69-77.
https://doi.org/10.1177/1082013207075465 |
| 55 | Qi X, Zhang Y, Guo H, Hai Y, Luo Y, Yue T: Mechanism and intervention measures of iron side effects on the intestine. Crit Rev Food Sci Nutr 2020;60:2113-2125.
https://doi.org/10.1080/10408398.2019.1630599 |
| 56 | Knutson MD, Walter PB, Ames BN, Viteri FE: Both iron deficiency and daily iron supplements increase lipid peroxidation in rats. J Nutr 2000;130:621-628.
https://doi.org/10.1093/jn/130.3.621 |
| 57 | King SM, Donangelo CM, Knutson MD, Walter PB, Ames BN: Daily supplementation with iron increases lipid peroxidation in young women with low iron stores. Exp Biol Med 2008;233:701-707.
https://doi.org/10.3181/0708-RM-233 |
| 58 | Xu S, He Y, Lin L, Chen P, Chen M, Zhang S: The emerging role of ferroptosis in intestinal disease. Cell Death Dis 2021;12:289.
https://doi.org/10.1038/s41419-021-03559-1 |
| 59 | Carrier JC, Aghdassi E, Jeejeebhoy K, Allard JP: Exacerbation of dextran sulfate sodium-induced colitis by dietary iron supplementation: role of NF-κB. Int J Colorectal Dis 2006;21:381-387.
https://doi.org/10.1007/s00384-005-0011-7 |
| 60 | Horniblow RD, Pathaka P, Balaccob DL, Acharjee A, Lles E, Gkoutos G, Beggs AD, Tselepis C: Iron-mediated epigenetic activation of NRF2 targets. J Nutr Biochem 2022;101:1-13.
https://doi.org/10.1016/j.jnutbio.2021.108929 |
| 61 | Kapoor B, Kapoor D, Gautam S, Singh R, Bhardwaj S: Dietary Polyunsaturated Fatty Acids (PUFAs): Uses and Potential Health Benefits. Curr Nutr Rep 2021;10:232-242.
https://doi.org/10.1007/s13668-021-00363-3 |
| 62 | Carvalho CCCR, Caramujo MJ: The Various Roles of Fatty Acids. Molecules 2018;23:1-36.
https://doi.org/10.3390/molecules23102583 |
| 63 | Gorjao R, Azevedo-Martins AK, Rodrigues HG, Abdulkader F, Arcisio-Miranda M, Procopio J, Curi R: Comparative effects of DHA and EPA on cell function. Pharmacol Ther 2009;122:56-64.
https://doi.org/10.1016/j.pharmthera.2009.01.004 |
| 64 | Goel A, Pothineni NV, Singhal M, Paydak H, Saldeen T: Fish, fish oils and cardioprotection: promise or fish tale? Int J Mol Sci 2018;19:1-13.
https://doi.org/10.3390/ijms19123703 |
| 65 | Shahidi F, Ambigaipalan P: Omega-3 polyunsaturated fatty acids and their health benefits. Annu Rev Food Sci Technol 2018;9:345-381.
https://doi.org/10.1146/annurev-food-111317-095850 |
| 66 | Food and Agriculture Organization of The United Nations Internet: Fats and fatty acids in human nutrition. Report of an expert consultation. FAO Food Nutr Pap 2010;91:1-166.
URL: https://www.fao.org/fileadmin/user_upload/nutrition/docs/requirements/fatsandfattacidsreport.pdf |
| 67 | Mills DE, William RG, Dixon H: Effects of dietary fatty-acid supplementation on fatty-acid composition and deformability of Young and old erythrocytes. Biochim Biophys Acta 1993;1149:313-318.
https://doi.org/10.1016/0005-2736(93)90216-M |
| 68 | Calder PC: n-3 fatty acids, inflammation and immunity: new mechanisms to explain old actions. Proc Nutr Soc 2013;72:326-36.
https://doi.org/10.1017/S0029665113001031 |
| 69 | Djuricic I, Calder PC: Beneficial Outcomes of Omega-6 and Omega-3 Polyunsaturated Fatty Acids on Human Health: An Update for 2021. Nutrients 2021;13:1-23.
https://doi.org/10.3390/nu13072421 |
| 70 | Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan WQ, Li P, Lu WJ, Watkins SM, Olefsky JM: GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010;142:687-698.
https://doi.org/10.1016/j.cell.2010.07.041 |
| 71 | Golpour P, Nourbakhsh M, Mazaherioun M, Janani L, Nourbakhsh M, Yaghmaei P: Improvement of NRF2 gene expression and antioxidant status in patients with type 2 diabetes mellitus after supplementation with omega-3 polyunsaturated fatty acids: A double-blind randomized placebo-controlled clinical Trial. Diabetes Res Clin Pract 2020;162:1-9.
https://doi.org/10.1016/j.diabres.2020.108120 |
| 72 | Das, UM: Saturated FattyAcids, MUFAs and PUFAs Regulate Ferroptosis. Cell Chem Biol 2019;26:309-311.
https://doi.org/10.1016/j.chembiol.2019.03.001 |
| 73 | Wong-Ekkabut J, Xu Z, Triampo W, Tang IM, Tieleman DP: Effect of lipid peroxidation on the properties of lipid bilayers: a molecular dynamics study. Biophys J 2007;93:4225-4236.
https://doi.org/10.1529/biophysj.107.112565 |
| 74 | Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W: Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015;520:57-62.
https://doi.org/10.1038/nature14344 |
| 75 | Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, Scott JP,Cai KQ, Campbell MR, Porter DK, Wang X, Bell DA, Li X, Garlick DS, Liu Q, Hollstein M, George DL, Murphy ME: An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev 2016;30:918-930.
https://doi.org/10.1101/gad.275891.115 |
| 76 | Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, Olzmann JA, Dixon SJ: Exogenous monounsaturated fatty acids promote a ferroptosis‐resistant cell state. Cell Chem Biol 2019;26:420-432.
https://doi.org/10.1016/j.chembiol.2018.11.016 |
| 77 | Bi J, Ichu TA, Zanca C, Yang H, Zhang W, Gu Y, Chowdhry S, Reed A, Ikegami S, Turner KM, Zhang W, Villa GR, Wu S, Quehenberger O, Yong WH, Kornblum HI, Rich JN, Cloughesy TF, Cavenee WK, Furnari FB, Cravatt BF, Mischel PS: Oncogene amplification in growth factor signaling pathways renders cancers dependent on membrane lipid remodeling. Cell Metab 2019;30:525-538.
https://doi.org/10.1016/j.cmet.2019.06.014 |
| 78 | Gimple RC, Kidwell RL, Kim LJY, Sun T, Gromovsky AD, Wu Q, Wolf M, Lv D, Bhargava S, Jiang L, Prager BC, Wang X, Ye Q, Zhu Z, Zhang G, Dong Z, Zhao L, Lee D, Bi J, Sloan AE, Mischel PS, Brown JM, Cang H, Huan T, Mack SC, Xie Q, Rich JN: Glioma Stem Cell-Specific Superenhancer Promotes Polyunsaturated Fatty-Acid Synthesis to Support EGFR Signaling. Cancer Discov 2019;9:1248-1267.
https://doi.org/10.1158/2159-8290.CD-19-0061 |
| 79 | de Assis AM, Rech A, Longoni A, Rotta LN, Denardin CC, Pasquali MA, Souza DO, Perry MLS, Moreira JC.: Ω3-Polyunsaturated fatty acids prevent lipoperoxidation, modulate antioxidant enzymes, and reduce lipid content but do not alter glycogen metabolism in the livers of diabetic rats fed on a high fat thermolyzed diet. Mol Cell Biochem 2012;361:151-160.
https://doi.org/10.1007/s11010-011-1099-4 |
| 80 | Giordano E, Visioli F: Long-chain omega 3 fatty acids: molecular bases of potential antioxidant actions. Prostaglandins Leukot Essent Fatty Acids 2014;90:1-4.
https://doi.org/10.1016/j.plefa.2013.11.002 |
| 81 | Mazereeuw G, Herrmann N, Andreazza AC, Scola G, Ma DWL, Oh PI, Lanctôt KL: Oxidative stress predicts depressive symptom changes with omega-3 fatty acid treatment in coronary artery disease patients. Brain Behav Immun 2017;60:136-141.
https://doi.org/10.1016/j.bbi.2016.10.005 |
| 82 | Allard JP, Kurian R, Aghdassi E, Muggli R, Royall D: Lipid peroxidation during n-3 fatty acid and vitamin E supplementation in humans. Lipids 1997;32:535-541.
https://doi.org/10.1007/s11745-997-0068-2 |
| 83 | Kesavulu MM, Kameswara Rao B, Apparao C, Kumar EGTV, Harinarayan CV: Effect of omega-3 fatty acids on lipid peroxidation and antioxidant enzyme status in type 2 diabetic patients. Diabetes Metab 2002;28:20-26. |
| 84 | Mansara P, Ketkar M, Deshpande R, Chaudhary A, Shinde K, Kaul-Ghanekar, R: Improved antioxidant status by omega-3 fatty acid supplementation in breast câncer patients undergoing chemotherapy: a case series. J Med Case Rep 2015;9:1-6.
https://doi.org/10.1186/s13256-015-0619-3 |
| 85 | Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D: Activation of the p62‐Keap1‐NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016;63:173-184.
https://doi.org/10.1002/hep.28251 |
| 86 | Zhang C, Yu JJ, Yang C, Shang S, Lv XX, Cui B, Hua F: Cross talk between ferroptosis and stress-Implications in cancer therapeutic responses. Cancer Innovation 2022;1:92-113.
https://doi.org/10.1002/cai2.7 |
| 87 | Rockfield S, Chhabra R, Robertson M, Rehman N, Bisht R, Nanjundan M: Links Between Iron and Lipids: Implications in Some Major Human Diseases. Pharmaceuticals 2018;11:1-14.
https://doi.org/10.3390/ph11040113 |
| 88 | Ogłuszka M, Lipiński P, Starzyński RR: Interaction between iron and omega-3 fatty acids metabolisms: where is the cross-link? Crit Rev Food Sci Nutr 2022;62:3002-3022.
https://doi.org/10.1080/10408398.2020.1862047 |
| 89 | Valenzuela R, Rincón-Cervera MA, Echeverría F, Barrera C, Espinosa A, Hernández-Rodas MC, Ortiz M, Valenzuela A, Videla LA: Iron-induced pro-oxidant and pro-lipogenic responses in relation to impaired synthesis and accretion of long-chain polyunsaturated fatty acids in rat hepatic and extrahepatic tissues. Nutrition 2018;45:49-58.
https://doi.org/10.1016/j.nut.2017.07.007 |
| 90 | Barrera C, Valenzuela R, Rincón MA, Espinosa A, López-Arana S, González-Mañan D, Romero N, Vargas R, Videla LA: Iron-induced derangement in hepatic Δ-5 and Δ-6 desaturation capacity and fatty acid profile leading to steatosis: Impact on extrahepatic tissues and prevention by antioxidant-rich extra virgin olive oil. Prostaglandins Leukot Essent Fatty Acids 2020;153:1-10.
https://doi.org/10.1016/j.plefa.2020.102058 |
| 91 | Bidault G, Virtue S, Petkevicius K, Jolin HE, Dugourd A, Guénantin AC, Leggat J, Mahler-Araujo B, Lam BYH, Ma MK, Dale M, Carobbio S, Kaser A, Fallon PG, Saez-Rodriguez J, McKenzie ANJ, Vidal-Puig A: SREBP1-induced fatty acid synthesis depletes macrophages antioxidant defenses to promote their alternative activation. Nat Metab 2021;3:1150-1162.
https://doi.org/10.1038/s42255-021-00440-5 |
| 92 | Xiaoli AM, Song Z, Yang, F: Lipogenic SREBP-1a/c transcription factors activate expression of the iron regulator hepcidin, revealing cross-talk between lipid and iron metabolisms. J Biol Chem 2019;294:12743-12753.
https://doi.org/10.1074/jbc.RA119.009644 |
| 93 | Ma Y, Smith CE, Lai CQ, Irvin MR, Parnell LD, Lee YC, Pham LD, Aslibekyan S, Claas SA, Tsai MY, Borecki IB, Kabagambe EK, Ordovás JM, Absher DM, Arnett DK: The effects of omega-3 polyunsaturated fatty acids and genetic variants on methylation levels of the interleukin-6 gene promoter. Molecular Nutrition & Food Research 2016;60:410-419.
https://doi.org/10.1002/mnfr.201500436 |
| 94 | Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, Ganz T: IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004;113:1271-1276.
https://doi.org/10.1172/JCI200420945 |