Unraveling the Connection Between Ion Channels and Pancreatic Stellate Cell Activation
Keywords
Abstract
Quiescent pancreatic stellate cells (PSCs) represent only a very low proportion of the pancreatic tissue, but their activation leads to stroma remodeling and fibrosis associated with pathologies such as chronic pancreatitis and pancreatic ductal adenocarcinoma (PDAC). PSC activation can be induced by various stresses, including acidosis, growth factors (PDGF, TGFβ), hypoxia, high pressure, or intercellular communication with pancreatic cancer cells. Activated PSC targeting represents a promising therapeutic strategy, but little is known regarding the molecular mechanisms underlying the activation of PSCs. Identification of new biomarkers of PSC activation associated with desmoplasia in chronic pancreatitis and PDAC could lead to new therapeutic targets for exocrine pancreatic disease treatments. Ion channels and transporters are transmembrane proteins involved in numerous physiological and pathological processes, including PDAC. They are well known to act as biosensors of the tissue microenvironment, and they can be easily accessible for drugs. However, their role in PSC activation is not fully understood. In this review, we briefly discuss the role of activated PSCs in pancreas inflammation and pathological fibrosis (associated with chronic pancreatitis and PDAC), and we describe the role of specific ion channels and transporters (Ca2+, K+, Na+ and Cl-) in these processes in the light of recent literature.Introduction
The pancreas is a digestive gland with two distinct physiological functions. The endocrine portion of the pancreas plays a pivotal role in maintaining optimal blood glucose levels through the production of pancreatic hormones, including insulin and glucagon, which are released into the bloodstream. The exocrine portion of the pancreas is responsible for the production and secretion of digestive enzymes and bicarbonate ions, which are transported through the pancreatic ducts to the duodenum, where they facilitate the completion of the digestive process. The exocrine part of the pancreatic tissue consists of acinar and ductal epithelial cells supported by a stroma containing blood vessels, cells and extracellular matrix (ECM). One of the distinctive characteristics of the pancreas, which it shares with the liver, is the presence of stellate cells within its stroma. Pancreatic stellate cells (PSCs) represent 4-7% of the healthy organ and are mainly involved in the regulation of tissue homeostasis by maintaining ECM turnover, but they also initiate pathological fibrosis when activated [1-4].
The pancreas is exposed to several stresses leading to tissue injury and acini degradation. Despite these injuries, pancreatic tissue can regenerate due to the plasticity of both epithelial and stellate cells. Acinar cells can adapt to their environment and response to stresses by transforming into duct or duct-like cells, a process called acinar-to-ductal metaplasia (ADM) [5, 6]. Metaplastic ductal-like cells have a proliferative phenotype which allows acinar repair and pancreatic tissue regeneration, avoiding self-digestion. ADM is under the control of RAS signaling, and it is also suggested as the first step of pancreatic carcinogenesis [7, 8]. Alongside ADM occurring in epithelial tissue, PSCs respond to injuries by activating. PSC activation leads to excess production of ECM inducing fibrosis. Both ADM and PSC activation are reversible, allowing pancreatic repair. However, chronic exposure to environmental stress can lead to pathological pancreatic remodeling and ultimately to neoplasia when the accumulation of mutations renders the remodeling irreversible [8]. The most common exocrine pancreatic diseases are pancreatitis and pancreatic ductal adenocarcinoma (PDAC, representing almost 90% of pancreatic cancers [8]). Chronic pancreatitis is defined as a pathologic fibro-inflammatory syndrome of the pancreas leading to progressive destruction of functional tissues [9]. The incidence of pancreatitis is generally low (4 to 14 cases for 100, 000 per year worldwide) but it is largely underestimated due to a lack of epidemiological studies [10]. Thus, recent studies suggest an increase in the incidence of chronic pancreatitis over the last 10 years [10]. Moreover, chronic pancreatitis increases risk of PDAC [11]. PDAC is the third-leading cause of mortality by cancer in men and women combined in the U.S., the seventh worldwide [8, 12], and it is expected to become the second one by 2040 [8, 13]. PDAC has a dramatically low 5-year overall survival rate of almost 10% [8, 14]. This poor outcome is explained by the fact that most patients are asymptomatic at early stages and present non-specific symptoms at advanced stages [8, 14, 15]. Consequently, PDAC is often diagnosed too late when the tumor is spread and unresectable. Moreover, PDAC is characterized by the formation of a fibrotic stroma with poor vascularization, which impedes the delivery of therapeutic molecules [8, 16]. This desmoplastic reaction is the primary cause of chemoresistance and therapeutic failure because the desmoplastic stroma establishes a barrier that prevents access to the drugs. Moreover, the desmoplastic stroma contains transformed cells (activated PSCs, cancer associated fibroblasts (CAFs), …) which communicate with pancreatic cancer cells by releasing tumor-promoting soluble factors (including growth factors) which stimulate chemoresistance as well as properties to form distant metastasis [1, 2, 4, 8, 17, 18]. The activated PSCs are known to be the initiators of both pancreatitis and PDAC desmoplasia [2-4]. The targeting of activated PSCs represents a promising therapeutic strategy that has attracted the attention of numerous research teams [1-3]. Thus, there is an urgent need to better understand the molecular mechanisms underlying the activation of PSCs leading to the desmoplastic reaction in order to propose new biomarkers and therapeutic targets of PDAC. Ion channels are plasma membrane proteins which are well known to act as biosensors of the tissue microenvironment. Ion channels are involved in cancer cell fates [19, 20]. Importantly, ion channels are located at the plasma membrane, and they can be easily accessible for drugs. Moreover, since numerous ion channel blockers have been already used for treating nervous and cardiovascular diseases, these drugs could be repurposed to selectively target ion channels in cancer [21]. Although the role of ion channels in cancer cells has been extensively documented, there is a paucity of research examining the significance of these proteins in PSC activation.
In this review, we briefly discuss the role of activated PSCs in pancreas inflammation and pathological fibrosis (associated with chronic pancreatitis and PDAC), and we describe the role of specific ion channels and transporters (Ca2+, K+, Na+ and Cl-) in these processes in the light of the literature.
Role of activated pancreatic stellate cells in pancreas inflammation and diseases
PSCs were first observed in mouse pancreas in 1982 [22]. They represent 4-7% of the healthy pancreas and are present in both endocrine (surrounding Langerhans islets) and exocrine (mainly surrounding the basolateral side of acinar cells but also surrounding small ducts) tissues. They are characterized by the presence of lipid droplets storing vitamin A in their cytosol and by the expression of markers such as desmin, glial fibrillary acidic protein (GFAP), nestin, neural cell adhesion molecule (NCAM), nerve growth factor (NGF), and synemin [23-25]. Despite the expression of neuroendocrine tissue-specific markers, a number of pieces of evidence support the hypothesis of mesenchymal, bone marrow cell and monocyte origins [3]. PSCs are classified into two distinct categories, quiescent and activated. Quiescent PSCs are important for pancreas regulation by maintaining ECM turnover, immune function, cell differentiation (such as hepatocytes, bile ductal and epithelial cells), and acetylcholine synthesis and secretion [4]. Upon activation, PSCs are no longer capable of storing vitamin A and undergo a loss of lipid droplets. They acquire a myofibroblast-like phenotype characterized by enhanced cell proliferation, migration and secretion. Activated PSCs express alpha smooth muscle actin (αSMA), fibroblast activation protein alpha (FAP-α), fibroblast specific protein-1 (FSP-1) and fibrinogen [18]. PSCs can be activated by different types of stress including ethanol and its metabolites, hypoxia, oxidative stress, pro-inflammatory cytokines (e.g., IL-1, IL-6, TNFα) and growth factors (platelet-derived growth factor (PDGF), transforming growth factor-β (TGF-β)) [18]. PSC activation is reversible and treatment with molecules such as troglitazone (a ligand of peroxisome proliferator activated receptor γ (PPAR-γ)), vitamin D analogues, and vitamin A (retinol) or its metabolites like all trans-retinoic acid (ATRA) can reprogram activated PSCs into a quiescent state [4]. Activated PSCs synthetize excessive ECM proteins as well as metalloproteases leading to a dense fibrotic stroma called desmoplasia [1, 4, 18]. Desmoplasia leads to fibrosis and pancreatitis. Importantly, the desmoplastic reaction gives rise to the formation of a hypovascularized, hypoxic, stiffer microenvironment which provides an optimal milieu for tumor development. In PDAC, the desmoplastic stroma accounts for 50 to 80% of the tumor and is correlated with a poor outcome [1, 8]. Moreover, activated PSCs are present in the desmoplastic stroma, but they are also the source of some of the CAFs, particularly myofibroblast-like (myCAFs) and inflammatory/secretory CAFs (iCAFs) [26]. However, a recent study suggests that PSC-derived CAFs represent only a minority of the CAF population in PDAC [27]. Given that PSC activation represents a key event in the initiation of the desmoplastic reaction, reprogramming activated PSCs into quiescent ones has been proposed as a promising strategy for combating PDAC. To date, the only clinical trial based on activated PSC reprograming is a phase I trial repurposing ATRA in combination with chemotherapy for PDAC treatment [28]. Recent advances have identified promising activated PSC targets including multiple ion channels and transporters. These latter could be exploited to develop new therapeutic strategies against PDAC.
Role of Ca2+ channels and transporters
Cytosolic calcium cation (Ca2+) is a ubiquitous second messenger involved in many physiological and pathological processes. The roles of Ca2+ in the physiology and the pathology of the exocrine pancreas have been extensively reviewed [29-31]. Notably, quiescent PSCs are sensitive to bradykinin (BK), a proinflammatory vasoactive nonapeptide which serum level increases during pancreatitis [29-33]. Physiological concentrations of BK (~50pM to 1nM) induces a biphasic Ca2+ response in rat PSCs through activation of the B2 receptors [31, 33]. The biphasic Ca2+ response is composed of a transient rise followed by a sustained phase of [Ca2+]i increase [31, 33]. The initial transient Ca2+ response to BK is due to G protein coupled receptor (GPCR)/phospholipase C (PLC) signaling cascade, leading to inositol trisphosphate receptor (IP3R) opening and Ca2+ release from endoplasmic reticular (ER) stores [31, 33]. The sustained phase of [Ca2+]i increase is dependent on extracellular Ca2+ influx through store-operated Ca2+ entry (SOCE) channels [31, 33]. SOCE represents a physiological process that serves to replenish Ca2+ supplies within ER lumen. Decrease of Ca2+ levels within ER lumen are sensed by the EF-hands of stromal interaction molecule 1 (STIM1) which activates, oligomerizes and translocates at junctions between ER and plasma membranes. At these junctions or puncta, STIM1 clusters bind and activate SOCE channels, leading to sustained Ca2+ influx [34]. SOCE channels have been identified as Ca2+ release-activated Ca2+ or CRAC channels, which main representatives are the Orai1 protein [35]. Both Orai1, Orai2 and STIM1 are expressed in mouse PSCs [36] and Orai1 inhibitor GSK-7975A prevents the plateau phase of [Ca2+]i rise but not the initial transient [33]. This increase of intracellular Ca2+ ([Ca2+]i) induced by BK leads to nitric oxide (NO) production by PSCs through Ca2+-sensitive enzyme nitric oxide synthase (NOS) [33]. However, the physiological role of BK-induced NO production by PSCs is not yet fully understood [29]. There is a considerable heterogeneity of Ca2+ response between the different types of pancreatic cells (e.g., acinar, neuron, macrophage and stellate) [29, 31]. PSC Ca2+ responses are not sensitive to membrane depolarization and PSCs do not seem to form functional synapses with pancreatic neurons, suggesting an absence of voltage-gated ion channels in normal PSCs [29].
As mentioned above, Ca2+ entry in PSCs is due to activation of SOCE channels after stimulation of GPCR, PLC signaling and Ca2+-release from ER stores. Radoslavova et al. showed that Orai1 channels contribute to SOCE in PS-1 pre-activated human PSCs and regulate cell proliferation and TGF-β secretion through Ca2+-dependent AKT signaling pathway [37]. Moreover, treatment of PS-1 cells with TGF-β stimulates Orai1 expression, SOCE, as well as Orai1-dependent cell proliferation and secretion suggesting a role of Orai1 in PSC activation induced by a TGF-β-dependent positive feedback autocrine loop [37]. Finally, collagen deposition from PS-1 cells stimulated by TGF-β and/or vitamin C is regulated by Orai1 modulation [38].
The superfamily of Transient Receptor Potential (TRP) channels is composed of various ion channels essential for cation fluxes and particularly for Ca2+ entries in non-excitable cells while they are also permeant to Na+, K+, Mg2+ and other metal ions such as Zn2+ [39-41]. TRP channels are involved in numerous physiological and pathological processes, and are considered as promising drug targets [42]. TRP channels are classified as ankyrin (TRPA), classical or canonical (TRPC), melastatin (TRPM), mucolipin (TRPML), NO-mechano-potential or NOMP (TRPN), polycystin (TRPP), and vanilloid (TRPV) based on their primary sequence [39-41]. TRP channels are activated by different stimuli including osmolarity, mechanical stretch, pH, temperature, … [39, 41]. As cellular sensors, TRP channels have been studied in PSCs because of the many environmental stresses to which the pancreas is subjected during the development of pathologies. For example, Fels et al. hypothesize that desmoplasia occurring during pancreatic carcinogenesis induces stroma rigidity and increased stiffness that could be sensed by PSCs through mechanosensitive ion channels [43]. They identify TRPC1, a Ca2+ permeable channel [44], as a part of the mechanism involved in PSC activation induced by elevated pressure in mice. It has been demonstrated that exposure to high pressure (100 mmHg, 24h) results in an increase of PSC migration and in an increase of constitutive cation entry which are both strongly attenuated in TRPC1 KO mice [43]. Furthermore, this has been confirmed by Radoslavova et al. by showing that TRPC1 regulates α-SMA expression and IL-6 secretion through the activation of the small and mothers against decapentaplegic homolog 2 (SMAD2) and the extracellular signal-regulated kinase (ERK1/2) pathways in human PSCs [45]. Moreover, Schwab’s group highlights another mechanism of Ca2+-regulated PSC migration involving the complex formed by Ca2+-activated K+ (KCa) and TRPC3 channels which mediate Ca2+ entry and calpain activation [46]. K+ channel opening induces membrane hyperpolarization, leading to enhanced electrochemical gradient for Ca2+ influx [47] through Ca2+ permeable channels such as TRPC3 [48]. By using data mining of published microarray analyses of microdissected patient tissue samples, Storck et al. showed that TRPC3 is mainly overexpressed in the stromal compartment of PDAC [46]. Moreover, they observed that almost two-thirds of TRPC3 are colocalized with KCa3.1 channels at the plasma membrane of RLT-PSCs, a human PSC model isolated from a chronic pancreatitis [46]. Finally, TRPC3 silencing abolishes PDGF-stimulated migration of RLT-PSCs and the Ca2+ response with no additional effect when combined with TRAM-34 KCa3.1 blocker, suggesting a functional interaction between the two channels [46]. The same group also studied the effects of hypoxia, another important feature of desmoplasia, on TRPC channel expression and PSC migration [49]. They identified TRPC6 as a regulator of 3D migration and constitutive cation entries stimulated by hypoxia (1% O2, 24h) in mouse PSCs [49]. Moreover, TRPC6 also regulates hypoxia-dependent secretions of cytokines that are involved in autocrine stimulation of PSCs [49]. Interestingly, both TRPC3 and TRPC6 are receptor-operated channels (ROC) which are activated following GPCR stimulation. These results suggest that ROC channels play an important role in the activation of PSCs mediated by soluble factors. TRPM7 is a dual function protein comprising a non-selective cation channel and an atypical kinase domain [50, 51]. TRPM7 is expressed ubiquitously but upregulated in PDAC [52, 53]. TRPM7 expression has been studied in hepatic stellate cells, where it regulates cell proliferation [54]. In human PSCs, we showed that TRPM7 expression depends on the state of activation [55]. TRPM7 expression is strongly decreased following treatment by ATRA while it increases in PSCs “educated” with media of pancreatic cancer cells, and also in RLT-PSCs [55]. TRPM7 regulates PSC proliferation through the stabilization of p53 via the PI3K/Akt pathway [55]. Moreover, data mining analysis shows that TRPM7 is upregulated in CAFs compared to fibroblasts from normal pancreas [55]. TRPV1, a thermosensitive channel activated by heat, mediates Ca2+ influx induced by hyperthermia [56] leading to heat shock factor 1 (HSF1) nuclear translocation and cell adaptation to stress [57]. Indeed, TRPV1 blockade enhances thermo-immunotherapy (use of local hyperthermia in combination with potent immunomodulator for treating neoplasia) by inhibition of TGFβ pathway leading to activation of immune response and notably reduced fibrosis in PDAC [57]. However, the role of TRPV1 in PSC activation is not yet fully understood.
Piezo channels have been identified as the main mechanoreceptors of cell membranes, as they allow cation (mostly Ca2+) entry following mechanical stimuli [58]. Interestingly, Romac et al. showed that Piezo1 is the main mechanosensitive ion channel expressed in the pancreas and its activation initiates pancreatitis in mice [59]. Mechanistically, Piezo1 activation is responsible for the initial Ca2+ rise which in turn activates TRPV4 leading to sustained Ca2+ increase, Ca2+ overload and trypsin release by acinar cells [60]. In a similar way, Swain et al. show that Piezo1 channels are expressed in PSCs and are responsible for pressure-induced fibrosis [61]. Mice where Piezo1 was deleted specifically in PSCs were protected against fibrosis induced by pancreatic duct ligation and intraductal elevated pressure [61]. Piezo1 activation by Yoda1 or by shear stress induces [Ca2+]i rise as well as fibrogenic response evaluated by fibronectin and collagen type I expression [61]. Finally, neither Yoda1 nor shear stress were able to induce sustained [Ca2+]i rise in PSCs isolated cells from TRPV4-KO mice, suggesting that TRPV4 opening is required to maintain Ca2+ overload induced by Piezo1 activation in PSCs exposed to high pressure [61]. Piezo1 channels are also involved in PSC migration, as demonstrated by Kuntze et al. [62]. More recently, Budde et al. showed that mechanosensitive channels including Piezo1, TRPV4 and TRPC1 are involved in PSC durotaxis which represents the directional cell migration along stiffness gradient of ECM [63]. In particular, they show that Piezo1 is essential for durotaxis but in interaction with TRPV4 or TRPC1 to allow an optimal PSC migration along stiffness gradient [63].
Nicotine, the major component of tobacco, is an independent risk factor of pancreatic fibrosis [64]. Li et al. showed that nicotine treatment (1µM, 48h) activates human PSCs leading to enhanced cell proliferation, reduced apoptosis, overexpression of α-SMA, secretion of ECM, and autophagy [64]. This work also shows that nicotine induced the overexpression of the α7 nicotinic acetylcholine receptor (α7nAChR), a ligand gated Ca2+ channel, in PSCs [64]. Use of the specific α7nAChR antagonist α-bungarotoxin (α-BTX, 0.1μM) inhibited the α7nAChR-mediated JAK2/STAT3 signaling pathway and prevented PSC activation induced by nicotine [64]. These results were confirmed in a rat model of chronic pancreatitis [65]. Interestingly, Wei et al. also demonstrated that nicotine induces fibrosis in rats and activates PSCs [66]. They further show that nicotine induces the production of reactive oxygen species (ROS) in the mitochondria as well as mitochondrial Ca2+ overload through upregulated mitochondrial Ca2+ uniporter (MCU) expression [66].
During pancreatic lesions associated with both pancreatitis and PDAC, ATP is released in stroma following acinar cell death [29]. It has been shown that extracellular ATP induces [Ca2+]i increase in mouse activated PSCs [67]. ATP regulates [Ca2+]i by binding to plasma membrane purinergic receptors such as P2X (ligand-gated Ca2+ channels) and P2Y (G-protein coupled receptors). Several P2X (P2X1, P2X4, P2X5, and P2X7) and P2Y (P2Y1, P2Y2, and P2Y6) receptors are expressed in PSCs [67-70]. Haanes et al. showed that P2X7 regulates mouse PSC proliferation in low serum conditions as well as [Ca2+]i [71]. Giannuzzo et al. showed also that PSCs isolated from P2X7-/- mice were unable to stimulate pancreatic cancer cell migration [72]. Moreover, treatment of orthotopic PancTu-1 Luc pancreatic tumor mouse model with the P2X7 inhibitor AZ10606120 reduces PSC activity and fibrosis [72]. In human RLT-PSCs, P2X7 stimulation by BzATP (an ATP analog which is 10-fold more potent than ATP to activate P2X7) induced [Ca2+]i increase, collagen secretion, and IL-6 release leading to stimulated pancreatic cancer cell migration through STAT3 pathway [73]. Interestingly, P2X7 variant polymorphisms with opposite roles have been described in PDAC tissues [74]. While the Gly150Arg variant expression has a protective effect on [Ca2+]i overload and cell migration, the Arg276His promotes [Ca2+]i increase and inflammatory cytokine release (IL-6, IL-1β, IL-8, TNF-α) as for the wild-type [74].
Intracellular Ca2+ can also be regulated by secondary active transport mechanisms such as the Na+/Ca2+ eXchanger (NCX) which allows Ca2+ extrusion (forward mode) or entry (reverse mode) depending on Na+ electrochemical gradient [75]. NCX1 is expressed in both murine and human PSCs [76]. In physiological conditions (pHe 7.4), NCX1 works in forward mode, prevents cellular Ca2+ accumulation, and slows down PSC migration [76]. Nevertheless, the role of NCX1 on PSC migration is dependent of tumoral microenvironment because NCX1 inhibition enhances PSC migration at pHe 7.4 while decreasing it at pHe 6.6 [76].
Taken together, these data show that PSC activation is highly regulated by Ca2+ toolkit depending on the environmental stimuli. The main results discussed in this section are summarized in Fig. 1.
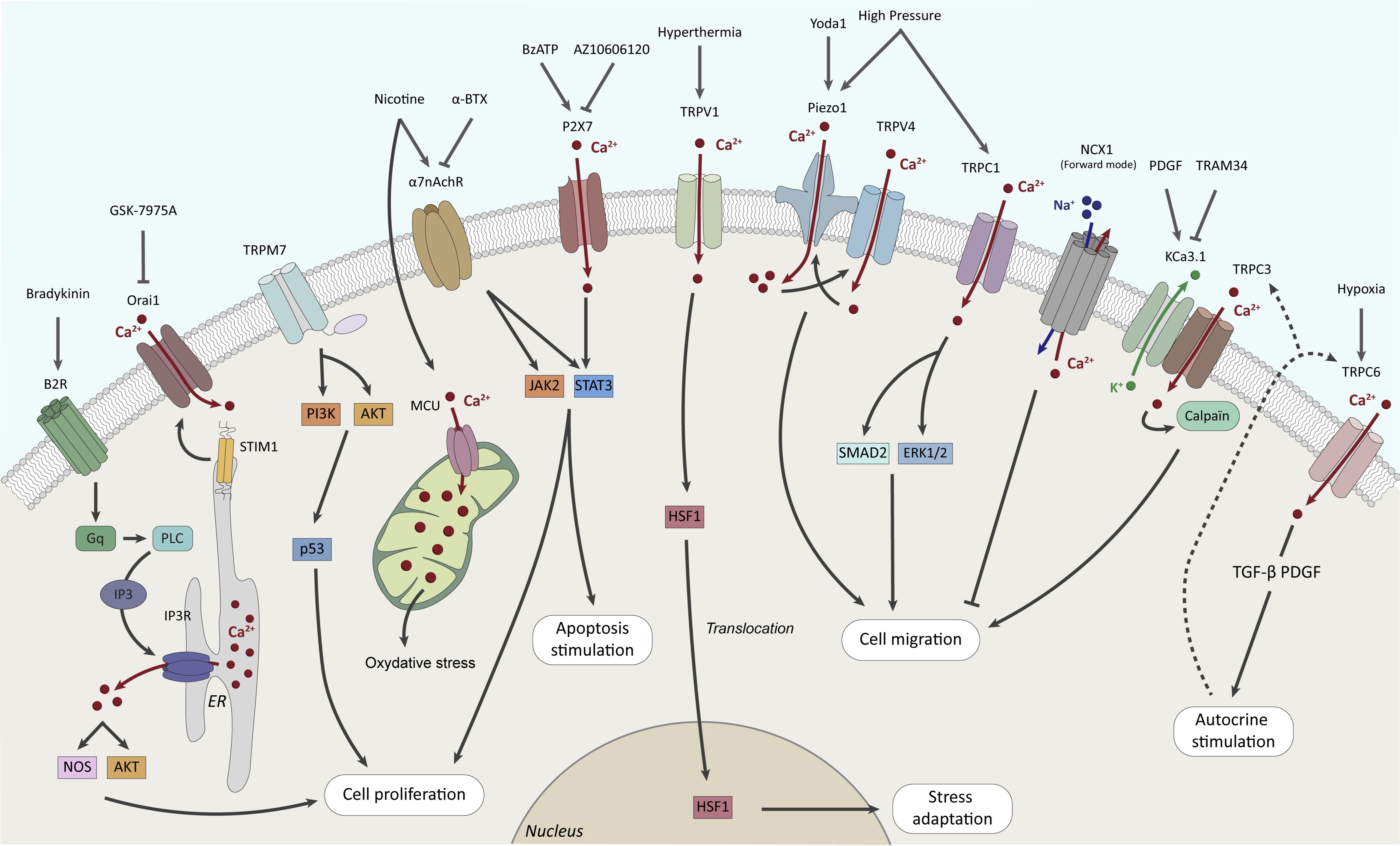
Fig. 1: Roles of Ca2+ channels and transporters in PSC fates.
Role of K+ channels and transporters
K+ is the most abundant cation in the cell. As mentioned above, PSCs express KCa channels [46] which are opened by intracellular Ca2+ increase and promote Ca2+ influx through Ca2+ channels (Orai or TRP channels) by hyperpolarizing the plasma membrane leading to a large electrochemical gradient for Ca2+ entry [29]. The K+ intermediate conductance Ca2+-activated KCa3.1 (also identified as KCNN4, IK1 or SK4) channels are expressed and functional at the plasma membrane of RLT-PSCs [46]. These channels are required for stimulated PSC migration through calpain activity and Ca2+ signaling by interacting with TRPC3 channels [46]. One of the major roles of PSCs in PDAC is the intercellular communication with cancer cells, leading to chemoresistance and metastasis. Interestingly, Rapetti-Mauss et al. described that the K+ small conductance Ca2+-activated SK2 channels expressed in the plasma membrane of pancreatic cancer cells are activated by the secretome of CAFs [77]. Importantly, SK2 is a part of a signaling complex, including Sigma1R (Sig-1R) chaperone, β1-integrin, EGFR and AKT, which can be targeted by Sig-1R ligand leading to delayed tumor progression and extended overall survival in an endogenous PDAC mice model [77]. While such mechanism has not been yet described in PSCs, it is tempting to speculate that KCa channels could also interact with other signaling proteins to regulate PSC activation and pancreatic fibrosis. Recently, K2P2.1 channels have been identified in murine PSCs where they regulate resting membrane potential and cell migration modulated by acidic extracellular pH (pHe 6.6) and high pressure (+100 mmHg) mimicking PDAC microenvironment [78].
The H+, K+-ATPase is expressed in normal pancreatic ductal cells where it regulates the intracellular K+ homeostasis, participates in the secretin-stimulated secretion, and in both intracellular (pHi) and extracellular pH (pHe) regulation by inducing active influx of K+ and outflow of H+ [79]. One of the hallmarks of PDAC is its acidic microenvironment which promotes fibrosis, resistance and metastasis [79, 80]. Tozzi et al. demonstrated that H+, K+-ATPases are expressed in RLT-PSCs at the leading edge of migrating cells [81]. Moreover, inhibition of these pumps by using selective blockers such as pantoprazole decreased PSC proliferation and collagen I secretion [81]. The main results discussed in this section are summarized in Fig. 2.
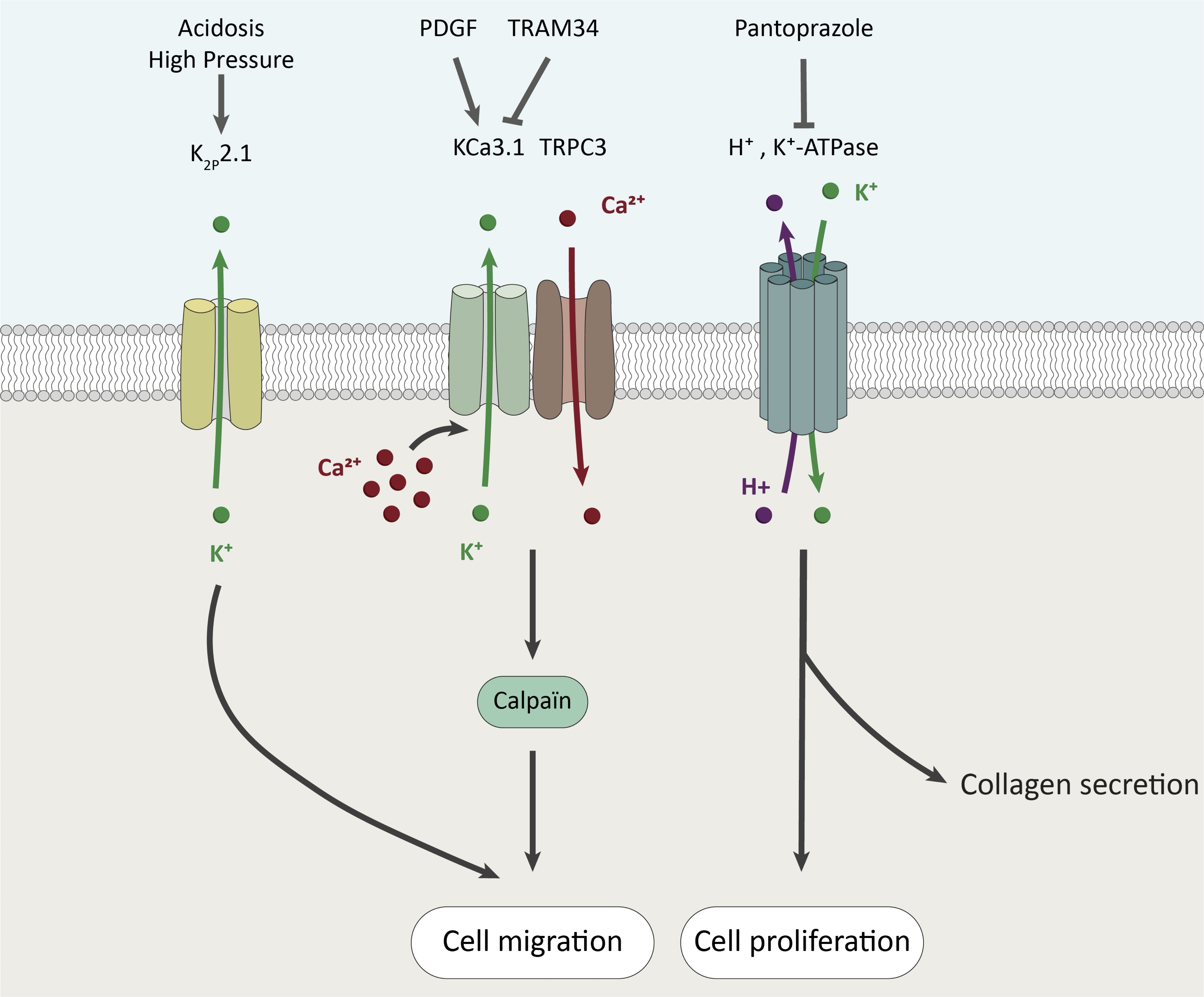
Fig. 2: Roles of K + channels and transporters in PSC fates.
Role of Na+ channels and transporters
Na+ is the most abundant cation of the extracellular fluids. In mouse and human PSCs, bile acid salts induce [Ca2+]i oscillations and necrosis which are dependent on extracellular Na+ [82]. The effect of bile acid salts on PSCs could be explained at least in part by the presence of the sodium–taurocholate cotransporting polypeptide (NTP) coding by the slc10A1 gene in both mouse and human PSCs [82]. As mentioned before, the Na+/Ca2+ eXchanger 1 (NCX1) regulates differently the PSC migration according to the tumor microenvironment, and particularly depending on the pH context [76]. Pancreatitis and PDAC development are accompanied by dramatic alterations of pH [79]. Several Na+ channels/transporters including Acid-Sensing Ion Channels (ASIC) and Na+/H+ Exchangers (NHE) are regulated by pH. ASIC1 channels have been detected in PSCs and their expression is upregulated by acidosis [83] but also by the co-culture with pancreatic cancer cells leading to PSC activation [84]. Interestingly, ASIC1 silencing in PSCs inhibited both cell viability and migration stimulated by conditioned media from pancreatic cancer cells [84]. Mechanistically, pancreatic cancer cells stimulate PSC viability and migration through ASIC1 and ERK signaling, but the role of Na+ influx and/or membrane depolarization still needs to be deciphered [84]. Moreover, Wang et al. showed that acidosis-induced PSC activation and autophagy, depend on ASIC1a channel activity and expression [83]. However, outside-out and whole-cell patch-clamp experiments in oocytes expressing ASIC1 showed that acidic pHe rapidly inactivates ASIC1 channels and the pHe must be returned to pHe 7.4 to reactivate the channels [85]. This suggests that ASIC1 activation by acidic pHe is probably transient, or there may be other factors present in the tumor microenvironment capable of permanently activating these channels. Pethő et al. described the role of NHE in PSC activation into a myofibroblast-like phenotype in vitro and in PDAC-associated fibrosis in vivo [86]. PSCs exposed to alkaline pHe (7.4 instead of 6.6) express myofibroblast-like markers, suggesting that the disruption of intermittent acidosis in the stroma induces PSC activation. These data support the hypothesis that PSCs are intermittently exposed to acidosis due to secretions of bicarbonates (HCO3-) in the pancreatic ducts (at the apical side of the plasma membrane) which are associated to a secretion of H+ in the stroma during the digestive processes [79, 86]. Thus, intermittent acidosis maintains the PSCs in a quiescent state. NHE1 encoded by the SLC9A1 gene is overexpressed in PSCs cultivated at pHe 7.4 and in PDAC-derived CAFs [86]. Moreover, NHE1 is mainly located at the plasma membrane and acts as a H+ extruder induced by Na+ influx in PSCs and PDAC-derived CAFs [86]. Finally, NHE1 activity maintains myofibroblast-like phenotype in PSCs exposed to acidic tumor microenvironment and NHE1 inhibition by cariporide can be used as a neoadjuvant drug in combination with gemcitabine to reduce fibrosis and improve the immune response [86]. The main results discussed in this section are summarized in Fig. 3.
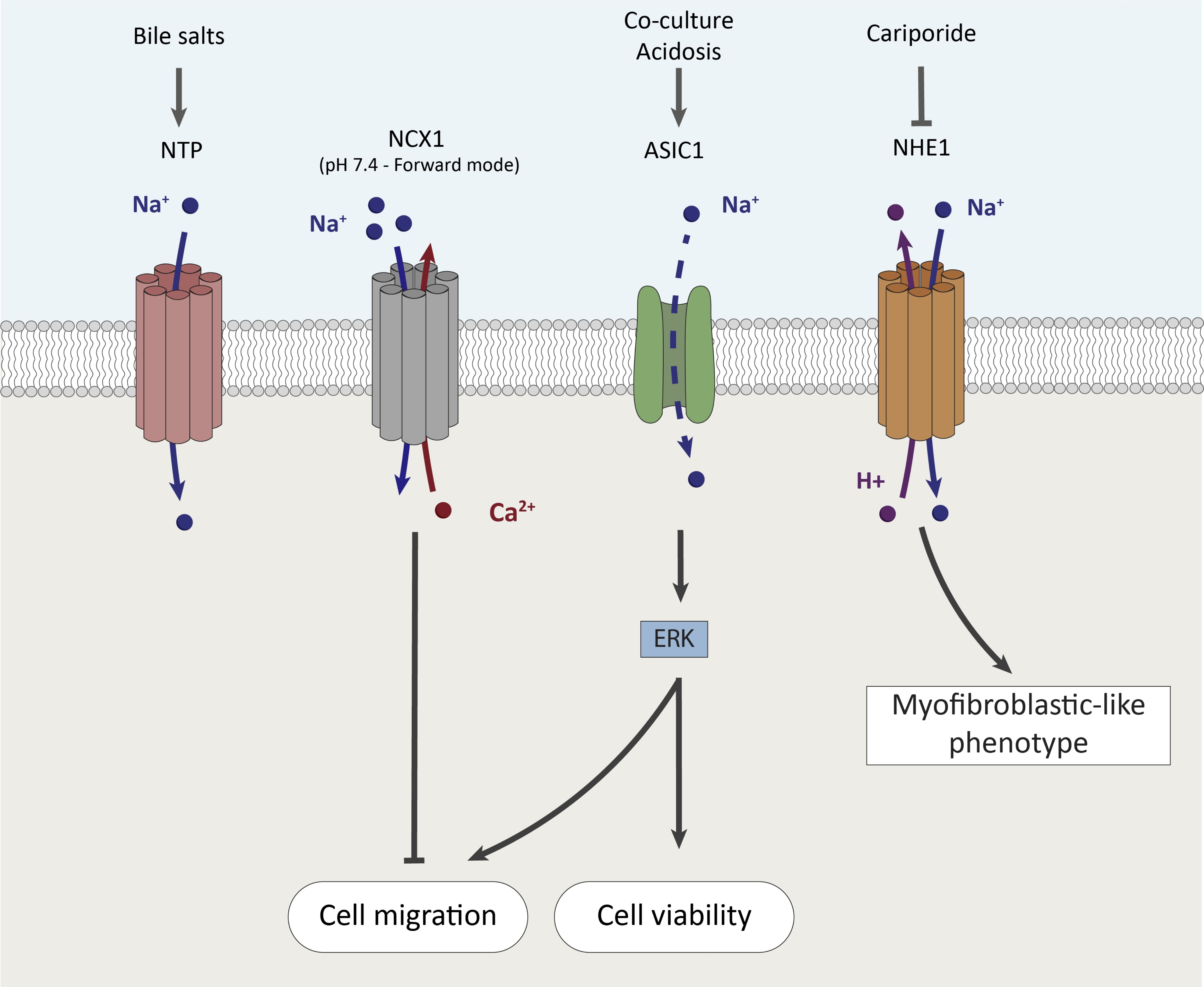
Fig. 3: Roles of Na+ channels and transporters in PSC fates.
Role of Cl- channels
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) is a chloride (Cl-) channel essential for fluid and electrolyte secretions in respiratory and digestive tracts. In the pancreas, CFTR is expressed in the apical membrane of centroacinar and proximal ductal cells, where it regulates ductal bicarbonate secretion. The mutation of the CFTR gene is associated with cystic fibrosis (CF) which can lead to abnormal function of the pancreas resulting in diabetes for almost half of the CF patients [87]. Moreover, CFTR mutation associated with mutations of other genes are frequently found in chronic pancreatitis [88]. In CFTR knockout animal models, it has been shown that CF is associated with activation of PSCs [87]. Moreover, the development of fibrosis in CFTR-/- sheep during gestation is accompanied by an enrichment of the stellate cell population in the pancreas [89]. However, while PSC abundance and activation were dependent on CFTR expression, a possible link between CFTR activity, Cl- flows and PSC activation has not yet been demonstrated. Moreover, it is possible that other Cl- channels are involved in PSC activation. For example, in human hepatic stellate cells, non-specific blockers (4, 40-diisothiocyanatostilbene-2, 20-disulfonic acid (DIDS), 5-nitro-2-(3-phenylpropyl-amino) benzoic acid (NPPB), and indanyloxyacetic acid (IAA-94)) of Cl- channels inhibited cell activation induced by reactive oxygen species [90].
Roles of metals?
Auwercx et al. showed that TRPM7 regulates human PSC proliferation through PI3K/Akt/p53 signaling pathway in a Mg2+-dependent manner [55]. Epidemiological studies show that low Mg2+ intake (<75% of recommendary daily allowance: ~219.43mg per day) is an independent risk factor of PDAC [91], and a low Mg2+ level is found in the serum of PDAC patients (0.72±0.13mM) compared to healthy controls (0.82±0.13mM) [92]. But the data published in cell and in animal models are often contradictory regarding the role of Mg2+ intake. Thus, low Mg2+ intake (from 1000 to 30mg/kg in diet) decreases lung cancer cell proliferation and primary tumor growth, while it promotes metastasis in mice [93]. Importantly, low Mg2+ (0.08mM) in the tumor microenvironment of pancreatic cancer cells potentiates allosteric inhibition of wild-type isocitrate dehydrogenase 1 (wtIDH1), improving survival in preclinical models [94]. Cellular Mg2+ homeostasis is complex and involves many channels and transporters [95]. To our knowledge, little is known about the regulation of Mg2+ in PSCs and further investigations are needed to better understand the role of Mg2+ in PSC activation and fibrosis.
There is growing evidence that trace metal exposure is associated with various pathologies, including cancer [96]. Recent data highlight an increase of toxic metal (i.e., As, Pb and Hg) concentrations in PDAC tissues compared to non-cancer adjacent ones [92]. However, the link between trace metal exposure and PSC activation has not yet been identified.
Future directions
In this review, we summarized the recent findings on ion channels associated with PSC activation, fibrosis and desmoplasia. Activation of PSCs can be induced by many stimuli, leading to fibrosis and progression of pancreatic diseases such as chronic pancreatitis or PDAC. The present review highlights a variety of mechanisms modulating the properties of PSCs involving ion channels. This can be partly explained by differences in the cell models used. The studies presented use PSCs from mouse models or human cell lines isolated from pancreatitis (RLT-PSC) or healthy pancreas (PS-1). Given the anatomical differences between the mouse and human pancreas, it is possible that there are also differences in the stroma and nature of the PSCs [97]. Furthermore, there is anatomical heterogeneity within the human pancreas and also in the types of PSCs that are activated [98]. Activation of PSCs can lead to the presence of several subpopulations of CAFs in the tumor microenvironment, including myCAF and secretory iCAF subtypes [99, 100]. It is not yet fully understood whether these phenotypes are a consequence of the different stimuli to which PSCs are exposed, or whether they are due to the origin of the PSCs. PSCs can be derived from different cell types, in particular hematopoietic stem cells. It is possible that their origin determines their activation mechanisms, their role in desmoplasia and tumor development, and probably the nature of the ion channels they express. It is important to be able to standardize the cellular models used to study PSCs and to better understand the origin and heterogeneity of these cells. To the best of our knowledge, no studies have yet been conducted on the expression or activity of ion channels in patient-derived CAFs. A systematic study of the ion channels expressed in primary CAFs as a function of clinical parameters could prove invaluable in understanding their role in carcinogenesis and, in particular, intercellular communication with cancer cells. Indeed, one of the defining characteristics of PDAC is the interaction between CAFs and cancer cells, which promotes metastasis formation and resistance to treatment. It is established that ion channels are involved in the activation of PSCs and in the regulation of their proliferation and migration. However, it has yet to be fully established if certain ion channels are involved in the ability of CAFs to stimulate pancreatic cancer cells.
Conclusion
In conclusion, recent studies have underscored the significance of ion channels in activating PSCs and regulating their characteristics. These channels can, therefore, be regarded as biomarkers of desmoplasia and as potential therapeutic targets. However, the heterogeneity of the pancreas, the cell models employed, and the subtypes of activated PSCs and CAFs imply that a more profound comprehension of these cells and the intricacy of their interactions with cancer cells is imperative.
Acknowledgements
Author Contributions
JA
designed and prepared the figures and corrected the manuscript. MF
and AL helped to write and corrected the manuscript. MG wrote the
manuscript and coordinated this work.
Funding Sources
Dr.
Julie Auwercx is founded by the Cancéropôle Nord-Ouest (CNO).
Mathilde Fourgeaud’s PhD is funded by The French Agency for
Ecological Transition (ADEME) and by l’Université de Picardie
Jules Verne (UPJV). Alexis Lalot’s PhD is funded by l’UPJV. This
work has been founded by The French National Research Program for
Environmental and Occupational Health of Anses. It has financial
support from ITMO Cancer of Aviesan on funds administered by Inserm
(2020/01/195), by the CNO, by the Ligue Contre le Cancer (Septentrion
and Somme’s committees), and by the MOSOPS project. The MOSOPS
project has received financial support from the French State,
Hauts-de-France region, INSERM and A2U Alliance’s Universities.
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors have no conflicts of interest to declare.
Disclosure of AI
AI tools have not been used to create this work.
References
| 1 | Chakkera M, Foote JB, Farran B, Nagaraju GP: Breaking the stromal barrier in pancreatic cancer: Advances and challenges. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 2024;1879:189065.
https://doi.org/10.1016/j.bbcan.2023.189065 |
| 2 | Pothula SP, Pirola RC, Wilson JS, Apte MV: Pancreatic stellate cells: Aiding and abetting pancreatic cancer progression. Pancreatology 2020;20:409-418.
https://doi.org/10.1016/j.pan.2020.01.003 |
| 3 | Hrabák P, Kalousová M, Krechler T, Zima T: Pancreatic stellate cells - rising stars in pancreatic pathologies. Physiological Research 2021:S597-S616.
https://doi.org/10.33549/physiolres.934783 |
| 4 | Sarkar R, Xu Z, Perera CJ, Apte MV: Emerging role of pancreatic stellate cell-derived extracellular vesicles in pancreatic cancer. Seminars in Cancer Biology 2023;93:114-122.
https://doi.org/10.1016/j.semcancer.2023.05.007 |
| 5 | Jiang T, Wei F, Xie K: Clinical significance of pancreatic ductal metaplasia. J Pathol 2022;257:125-139.
https://doi.org/10.1002/path.5883 |
| 6 | Backx E, Coolens K, Van Den Bossche J-L, Houbracken I, Espinet E, Rooman I: On the Origin of Pancreatic Cancer: Molecular Tumor Subtypes in Perspective of Exocrine Cell Plasticity. Cellular and Molecular Gastroenterology and Hepatology 2022;13:1243-1253.
https://doi.org/10.1016/j.jcmgh.2021.11.010 |
| 7 | Giroux V, Rustgi AK: Metaplasia: tissue injury adaptation and a precursor to the dysplasia-cancer sequence. Nature Reviews Cancer 2017;17:594-604.
https://doi.org/10.1038/nrc.2017.68 |
| 8 | Halbrook CJ, Lyssiotis CA, Pasca di Magliano M, Maitra A: Pancreatic cancer: Advances and challenges. Cell 2023;186:1729-1754.
https://doi.org/10.1016/j.cell.2023.02.014 |
| 9 | Whitcomb DC, Frulloni L, Garg P, Greer JB, Schneider A, Yadav D, Shimosegawa T: Chronic pancreatitis: An international draft consensus proposal for a new mechanistic definition. Pancreatology 2016;16:218-224.
https://doi.org/10.1016/j.pan.2016.02.001 |
| 10 | Kleeff J, Whitcomb DC, Shimosegawa T, Esposito I, Lerch MM, Gress T, Mayerle J, Drewes AM, Rebours V, Akisik F, Munoz JED, Neoptolemos JP: Chronic pancreatitis. Nat Rev Dis Primers 2017;3:17060.
https://doi.org/10.1038/nrdp.2017.60 |
| 11 | Kirkegård J, Mortensen FV, Cronin-Fenton D: Chronic Pancreatitis and Pancreatic Cancer Risk: A Systematic Review and Meta-analysis. American Journal of Gastroenterology 2017;112:1366-1372.
https://doi.org/10.1038/ajg.2017.218 |
| 12 | Siegel RL, Giaquinto AN, Jemal A: Cancer statistics, 2024. CA Cancer J Clin 2024;74:12-49.
https://doi.org/10.3322/caac.21820 |
| 13 | Rahib L, Wehner MR, Matrisian LM, Nead KT: Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw Open 2021;4:e214708.
https://doi.org/10.1001/jamanetworkopen.2021.4708 |
| 14 | Park W, Chawla A, O'Reilly EM: Pancreatic Cancer: A Review. Jama 2021;326:851-862.
https://doi.org/10.1001/jama.2021.13027 |
| 15 | Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, Neale RE, Tempero M, Tuveson DA, Hruban RH, Neoptolemos JP: Pancreatic cancer. Nat Rev Dis Primers 2016;2:16022.
https://doi.org/10.1038/nrdp.2016.22 |
| 16 | Aiello NM, Bajor DL, Norgard RJ, Sahmoud A, Bhagwat N, Pham MN, Cornish TC, Iacobuzio-Donahue CA, Vonderheide RH, Stanger BZ: Metastatic progression is associated with dynamic changes in the local microenvironment. Nature communications 2016;7:12819.
https://doi.org/10.1038/ncomms12819 |
| 17 | Rebelo R, Xavier CPR, Giovannetti E, Vasconcelos MH: Fibroblasts in pancreatic cancer: molecular and clinical perspectives. Trends Mol Med 2023;29:439-453.
https://doi.org/10.1016/j.molmed.2023.03.002 |
| 18 | Fu Y, Liu S, Zeng S, Shen H: The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma. Molecular Cancer 2018;17.
https://doi.org/10.1186/s12943-018-0815-z |
| 19 | Hofschröer V, Najder K, Rugi M, Bouazzi R, Cozzolino M, Arcangeli A, Panyi G, Schwab A: Ion Channels Orchestrate Pancreatic Ductal Adenocarcinoma Progression and Therapy. Frontiers in Pharmacology 2021;11.
https://doi.org/10.3389/fphar.2020.586599 |
| 20 | Prevarskaya N, Skryma R, Shuba Y: Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiological Reviews 2018;98:559-621.
https://doi.org/10.1152/physrev.00044.2016 |
| 21 | Kofman K, Levin M: Bioelectric pharmacology of cancer: A systematic review of ion channel drugs affecting the cancer phenotype. Progress in Biophysics and Molecular Biology 2024;191:25-39.
https://doi.org/10.1016/j.pbiomolbio.2024.07.005 |
| 22 | Watari N, Hotta Y, Mabuchi Y: Morphological studies on a vitamin A-storing cell and its complex with macrophage observed in mouse pancreatic tissues following excess vitamin A administration. Okajimas Folia Anat Jpn 1982;58:837-858.
https://doi.org/10.2535/ofaj1936.58.4-6_837 |
| 23 | Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, Pirola RC, Wilson JS: Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 1998;43:128-133.
https://doi.org/10.1136/gut.43.1.128 |
| 24 | Froeling FE, Mirza TA, Feakins RM, Seedhar A, Elia G, Hart IR, Kocher HM: Organotypic culture model of pancreatic cancer demonstrates that stromal cells modulate E-cadherin, beta-catenin, and Ezrin expression in tumor cells. The American journal of pathology 2009;175:636-648.
https://doi.org/10.2353/ajpath.2009.090131 |
| 25 | Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, Siech M, Beger H, Grunert A, Adler G: Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998;115:421-432.
https://doi.org/10.1016/S0016-5085(98)70209-4 |
| 26 | Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, Tuveson DA: IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discovery 2019;9:282-301.
https://doi.org/10.1158/2159-8290.CD-18-0710 |
| 27 | Helms EJ, Berry MW, Chaw RC, Dufort CC, Sun D, Onate MK, Oon C, Bhattacharyya S, Sanford-Crane H, Horton W, Finan JM, Sattler A, Makar R, Dawson DW, Xia Z, Hingorani SR, Sherman MH: Mesenchymal Lineage Heterogeneity Underlies Nonredundant Functions of Pancreatic Cancer-Associated Fibroblasts. Cancer Discovery 2022;12:484-501.
https://doi.org/10.1158/2159-8290.CD-21-0601 |
| 28 | Kocher HM, Basu B, Froeling FEM, Sarker D, Slater S, Carlin D, Desouza NM, De Paepe KN, Goulart MR, Hughes C, Imrali A, Roberts R, Pawula M, Houghton R, Lawrence C, Yogeswaran Y, Mousa K, Coetzee C, Sasieni P, Prendergast A, Propper DJ: Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nature Communications 2020;11.
https://doi.org/10.1038/s41467-020-18636-w |
| 29 | Petersen OH, Gerasimenko JV, Gerasimenko OV, Gryshchenko O, Peng S: The roles of calcium and ATP in the physiology and pathology of the exocrine pancreas. Physiological Reviews 2021;101:1691-1744.
https://doi.org/10.1152/physrev.00003.2021 |
| 30 | Gerasimenko JV, Peng S, Tsugorka T, Gerasimenko OV: Ca(2+) signalling underlying pancreatitis. Cell Calcium 2017.
https://doi.org/10.1016/j.ceca.2017.05.010 |
| 31 | Gerasimenko JV, Gerasimenko OV: The role of Ca2+ signalling in the pathology of exocrine pancreas. Cell Calcium 2023;112:102740.
https://doi.org/10.1016/j.ceca.2023.102740 |
| 32 | Hirata M, Hayashi I, Yoshimura K, Ishii KI, Soma K, Ohwada T, Kakita A, Majima M: Blockade of bradykinin B2 receptor suppresses acute pancreatitis induced by obstruction of the pancreaticobiliary duct in rats. British Journal of Pharmacology 2002;135:29-36.
https://doi.org/10.1038/sj.bjp.0704462 |
| 33 | Gryshchenko O, Gerasimenko JV, Gerasimenko OV, Petersen OH: Ca2+ signals mediated by bradykinin type 2 receptors in normal pancreatic stellate cells can be inhibited by specific Ca2+ channel blockade. The Journal of Physiology 2016;594:281-293.
https://doi.org/10.1113/JP271468 |
| 34 | Lur G, Haynes LP, Prior IA, Gerasimenko OV, Feske S, Petersen OH, Burgoyne RD, Tepikin AV: Ribosome-free Terminals of Rough ER Allow Formation of STIM1 Puncta and Segregation of STIM1 from IP3 Receptors. Current Biology 2009;19:1648-1653.
https://doi.org/10.1016/j.cub.2009.07.072 |
| 35 | Bakowski D, Murray F, Parekh AB: Store-Operated Ca(2+) Channels: Mechanism, Function, Pharmacology, and Therapeutic Targets. Annu Rev Pharmacol Toxicol 2021;61:629-654.
https://doi.org/10.1146/annurev-pharmtox-031620-105135 |
| 36 | Waldron RT, Chen Y, Pham H, Go A, Su HY, Hu C, Wen L, Husain SZ, Sugar CA, Roos J, Ramos S, Lugea A, Dunn M, Stauderman K, Pandol SJ: The Orai Ca2+ channel inhibitor CM4620 targets both parenchymal and immune cells to reduce inflammation in experimental acute pancreatitis. The Journal of Physiology 2019;597:3085-3105.
https://doi.org/10.1113/JP277856 |
| 37 | Radoslavova S, Folcher A, Lefebvre T, Kondratska K, Guenin S, Dhennin-Duthille I, Gautier M, Prevarskaya N, Ouadid-Ahidouch H: Orai1 Channel Regulates Human-Activated Pancreatic Stellate Cell Proliferation and TGFbeta1 Secretion through the AKT Signaling Pathway. Cancers (Basel) 2021;13.
https://doi.org/10.3390/cancers13102395 |
| 38 | Schleinhege R, Neumann I, Oeckinghaus A, Schwab A, Petho Z: A CNA-35-based high-throughput fibrosis assay reveals ORAI1 as a regulator of collagen release from pancreatic stellate cells. Matrix Biol 2024.
https://doi.org/10.1016/j.matbio.2024.12.004 |
| 39 | Du Y, Chen J, Shen L, Wang B: TRP channels in inflammatory bowel disease: Potential therapeutic targets. Biochem Pharmacol 2022;203:115195.
https://doi.org/10.1016/j.bcp.2022.115195 |
| 40 | Wu LJ, Sweet TB, Clapham DE: International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol Rev 2010;62:381-404.
https://doi.org/10.1124/pr.110.002725 |
| 41 | Zhang M, Ma Y, Ye X, Zhang N, Pan L, Wang B: TRP (transient receptor potential) ion channel family: structures, biological functions and therapeutic interventions for diseases. Signal Transduction and Targeted Therapy 2023;8.
https://doi.org/10.1038/s41392-023-01464-x |
| 42 | Koivisto A-P, Belvisi MG, Gaudet R, Szallasi A: Advances in TRP channel drug discovery: from target validation to clinical studies. Nature Reviews Drug Discovery 2022;21:41-59.
https://doi.org/10.1038/s41573-021-00268-4 |
| 43 | Fels B, Nielsen N, Schwab A: Role of TRPC1 channels in pressure-mediated activation of murine pancreatic stellate cells. European Biophysics Journal 2016;45:657-670.
https://doi.org/10.1007/s00249-016-1176-4 |
| 44 | Zitt C, Zobel A, Obukhov AG, Harteneck C, Kalkbrenner F, Luckhoff A, Schultz G: Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron 1996;16:1189-1196.
https://doi.org/10.1016/S0896-6273(00)80145-2 |
| 45 | Radoslavova S, Fels B, Pethö Z, Gruner M, Ruck T, Meuth SG, Folcher A, Prevarskaya N, Schwab A, Ouadid-Ahidouch H: TRPC1 channels regulate the activation of pancreatic stellate cells through ERK1/2 and SMAD2 pathways and perpetuate their pressure-mediated activation. Cell Calcium 2022;106:102621.
https://doi.org/10.1016/j.ceca.2022.102621 |
| 46 | Storck H, Hild B, Schimmelpfennig S, Sargin S, Nielsen N, Zaccagnino A, Budde T, Novak I, Kalthoff H, Schwab A: Ion channels in control of pancreatic stellate cell migration. Oncotarget 2017;8:769-784.
https://doi.org/10.18632/oncotarget.13647 |
| 47 | Mignen O, Constantin B, Potier-Cartereau M, Penna A, Gautier M, Gueguinou M, Renaudineau Y, Shoji KF, Felix R, Bayet E, Buscaglia P, Debant M, Chantome A, Vandier C: Constitutive calcium entry and cancer: updated views and insights. Eur Biophys J 2017.
https://doi.org/10.1007/s00249-017-1216-8 |
| 48 | Zitt C, Obukhov AG, Strubing C, Zobel A, Kalkbrenner F, Luckhoff A, Schultz G: Expression of TRPC3 in Chinese hamster ovary cells results in calcium-activated cation currents not related to store depletion. J Cell Biol 1997;138:1333-1341.
https://doi.org/10.1083/jcb.138.6.1333 |
| 49 | Nielsen N, Kondratska K, Ruck T, Hild B, Kovalenko I, Schimmelpfennig S, Welzig J, Sargin S, Lindemann O, Christian S, Meuth SG, Prevarskaya N, Schwab A: TRPC6 channels modulate the response of pancreatic stellate cells to hypoxia. Pflügers Archiv - European Journal of Physiology 2017;469:1567-1577.
https://doi.org/10.1007/s00424-017-2057-0 |
| 50 | Runnels LW, Yue L, Clapham DE: TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science 2001;291:1043-1047.
https://doi.org/10.1126/science.1058519 |
| 51 | Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg AM, Fleig A: LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 2001;411:590-595.
https://doi.org/10.1038/35079092 |
| 52 | Rybarczyk P, Gautier M, Hague F, Dhennin-Duthille I, Chatelain D, Kerr-Conte J, Pattou F, Regimbeau JM, Sevestre H, Ouadid-Ahidouch H: Transient receptor potential melastatin-related 7 channel is overexpressed in human pancreatic ductal adenocarcinomas and regulates human pancreatic cancer cell migration. Int J Cancer 2012;131:E851-861.
https://doi.org/10.1002/ijc.27487 |
| 53 | Yee NS, Kazi AA, Li Q, Yang Z, Berg A, Yee RK: Aberrant over-expression of TRPM7 ion channels in pancreatic cancer: required for cancer cell invasion and implicated in tumor growth and metastasis. Biology open 2015;4:507-514.
https://doi.org/10.1242/bio.20137088 |
| 54 | Rychkov GY, Barritt GJ: Expression and function of TRP channels in liver cells. Adv Exp Med Biol 2011;704:667-686.
https://doi.org/10.1007/978-94-007-0265-3_35 |
| 55 | Auwercx J, Kischel P, Lefebvre T, Jonckheere N, Vanlaeys A, Guenin S, Radoslavova S, Van Seuningen I, Ouadid-Ahidouch H, Kocher HM, Dhennin-Duthille I, Gautier M: TRPM7 Modulates Human Pancreatic Stellate Cell Activation. Cells 2022;11.
https://doi.org/10.3390/cells11142255 |
| 56 | Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D: The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997;389:816-824.
https://doi.org/10.1038/39807 |
| 57 | Li T, Jiang S, Zhang Y, Luo J, Li M, Ke H, Deng Y, Yang T, Sun X, Chen H: Nanoparticle-mediated TRPV1 channel blockade amplifies cancer thermo-immunotherapy via heat shock factor 1 modulation. Nature Communications 2023;14.
https://doi.org/10.1038/s41467-023-38128-x |
| 58 | Miyamoto T, Mochizuki T, Nakagomi H, Kira S, Watanabe M, Takayama Y, Suzuki Y, Koizumi S, Takeda M, Tominaga M: Functional role for Piezo1 in stretch-evoked Ca(2)(+) influx and ATP release in urothelial cell cultures. J Biol Chem 2014;289:16565-16575.
https://doi.org/10.1074/jbc.M113.528638 |
| 59 | Romac JM, Shahid RA, Swain SM, Vigna SR, Liddle RA: Piezo1 is a mechanically activated ion channel and mediates pressure induced pancreatitis. Nat Commun 2018;9:1715.
https://doi.org/10.1038/s41467-018-04194-9 |
| 60 | Swain SM, Romac JM, Shahid RA, Pandol SJ, Liedtke W, Vigna SR, Liddle RA: TRPV4 channel opening mediates pressure-induced pancreatitis initiated by Piezo1 activation. J Clin Invest 2020;130:2527-2541.
https://doi.org/10.1172/JCI134111 |
| 61 | Swain SM, Romac JM, Vigna SR, Liddle RA: Piezo1-mediated stellate cell activation causes pressure-induced pancreatic fibrosis in mice. JCI Insight 2022;7.
https://doi.org/10.1172/jci.insight.158288 |
| 62 | Kuntze A, Goetsch O, Fels B, Najder K, Unger A, Wilhelmi M, Sargin S, Schimmelpfennig S, Neumann I, Schwab A, Pethő Z: Protonation of Piezo1 Impairs Cell-Matrix Interactions of Pancreatic Stellate Cells. Frontiers in Physiology 2020;11.
https://doi.org/10.3389/fphys.2020.00089 |
| 63 | Budde I, Schlichting A, Ing D, Schimmelpfennig S, Kuntze A, Fels B, Romac JM, Swain SM, Liddle RA, Stevens A, Schwab A, Pethő Z: Piezo1-induced durotaxis of pancreatic stellate cells depends on TRPC1 and TRPV4 channels. bioRxiv 2024.
https://doi.org/10.1101/2023.12.22.572956 |
| 64 | Li Z, Zhang X, Jin T, Hao J: Nicotine promotes activation of human pancreatic stellate cells through inducing autophagy via α7nAChR-mediated JAK2/STAT3 signaling pathway. Life Sciences 2020;243:117301.
https://doi.org/10.1016/j.lfs.2020.117301 |
| 65 | Li Z, Lu D, Jin T, Liu X, Hao J: Nicotine facilitates pancreatic fibrosis by promoting activation of pancreatic stellate cells via α7nAChR-mediated JAK2/STAT3 signaling pathway in rats. Toxicology Letters 2021;349:84-91.
https://doi.org/10.1016/j.toxlet.2021.06.012 |
| 66 | Wei X, Yuan Y, Li M, Li Z, Wang X, Cheng H, Liu X, Hao J, Jin T: Nicotine aggravates pancreatic fibrosis in mice with chronic pancreatitis via mitochondrial calcium uniporter. Tob Induc Dis 2024;22.
https://doi.org/10.18332/tid/186587 |
| 67 | Won JH, Zhang Y, Ji B, Logsdon CD, Yule DI: Phenotypic changes in mouse pancreatic stellate cell Ca2+ signaling events following activation in culture and in a disease model of pancreatitis. Mol Biol Cell 2011;22:421-436.
https://doi.org/10.1091/mbc.e10-10-0807 |
| 68 | Burnstock G, Novak I: Purinergic signalling in the pancreas in health and disease. J Endocrinol 2012;213:123-141.
https://doi.org/10.1530/JOE-11-0434 |
| 69 | Hennigs JK, Seiz O, Spiro J, Berna MJ, Baumann HJ, Klose H, Pace A: Molecular basis of P2-receptor-mediated calcium signaling in activated pancreatic stellate cells. Pancreas 2011;40:740-746.
https://doi.org/10.1097/MPA.0b013e31821b5b68 |
| 70 | Kunzli BM, Berberat PO, Giese T, Csizmadia E, Kaczmarek E, Baker C, Halaceli I, Buchler MW, Friess H, Robson SC: Upregulation of CD39/NTPDases and P2 receptors in human pancreatic disease. Am J Physiol Gastrointest Liver Physiol 2007;292:G223-230.
https://doi.org/10.1152/ajpgi.00259.2006 |
| 71 | Haanes KA, Schwab A, Novak I: The P2X7 receptor supports both life and death in fibrogenic pancreatic stellate cells. PLoS One 2012;7:e51164.
https://doi.org/10.1371/journal.pone.0051164 |
| 72 | Giannuzzo A, Saccomano M, Napp J, Ellegaard M, Alves F, Novak I: Targeting of the P2X7 receptor in pancreatic cancer and stellate cells. Int J Cancer 2016;139:2540-2552.
https://doi.org/10.1002/ijc.30380 |
| 73 | Magni L, Bouazzi R, Heredero Olmedilla H, Petersen PSS, Tozzi M, Novak I: The P2X7 Receptor Stimulates IL-6 Release from Pancreatic Stellate Cells and Tocilizumab Prevents Activation of STAT3 in Pancreatic Cancer Cells. Cells 2021;10.
https://doi.org/10.3390/cells10081928 |
| 74 | Magni L, Yu H, Christensen NM, Poulsen MH, Frueh A, Deshar G, Johansen AZ, Johansen JS, Pless SA, Jorgensen NR, Novak I: Human P2X7 receptor variants Gly150Arg and Arg276His polymorphisms have differential effects on risk association and cellular functions in pancreatic cancer. Cancer Cell Int 2024;24:148.
https://doi.org/10.1186/s12935-024-03339-9 |
| 75 | Loeck T, Schwab A: The role of the Na+/Ca2+-exchanger (NCX) in cancer-associated fibroblasts. Biological Chemistry 2023;404:325-337.
https://doi.org/10.1515/hsz-2022-0253 |
| 76 | Loeck T, Rugi M, Todesca LM, Kalinowska P, Soret B, Neumann I, Schimmelpfennig S, Najder K, Pethő Z, Farfariello V, Prevarskaya N, Schwab A: The context-dependent role of the Na+/Ca2+-exchanger (NCX) in pancreatic stellate cell migration. Pflügers Archiv - European Journal of Physiology 2023;475:1225-1240.
https://doi.org/10.1007/s00424-023-02847-3 |
| 77 | Rapetti-Mauss R, Nigri J, Berenguier C, Finetti P, Tubiana SS, Labrum B, Allegrini B, Pellissier B, Efthymiou G, Hussain Z, Bousquet C, Dusetti N, Bertucci F, Guizouarn H, Melnyk P, Borgese F, Tomasini R, Soriani O: SK2 channels set a signalling hub bolstering CAF-triggered tumourigenic processes in pancreatic cancer. Gut 2023;72:722-735.
https://doi.org/10.1136/gutjnl-2021-326610 |
| 78 | Rugi M, Hofschroer V, Petho Z, Soret B, Loeck T, Schwab A: K(2P)2.1 channels modulate the pH- and mechanosensitivity of pancreatic stellate cells. Pflugers Arch 2024.
https://doi.org/10.1007/s00424-024-03021-z |
| 79 | Pedersen SF, Novak I, Alves F, Schwab A, Pardo LA: Alternating pH landscapes shape epithelial cancer initiation and progression: Focus on pancreatic cancer. Bioessays 2017;39.
https://doi.org/10.1002/bies.201600253 |
| 80 | Andersen HB, Ialchina R, Pedersen SF, Czaplinska D: Metabolic reprogramming by driver mutation-tumor microenvironment interplay in pancreatic cancer: new therapeutic targets. Cancer Metastasis Rev 2021;40:1093-1114.
https://doi.org/10.1007/s10555-021-10004-4 |
| 81 | Tozzi M, Sorensen CE, Magni L, Christensen NM, Bouazzi R, Buch CM, Stefanini M, Duranti C, Arcangeli A, Novak I: Proton Pump Inhibitors Reduce Pancreatic Adenocarcinoma Progression by Selectively Targeting H(+), K(+)-ATPases in Pancreatic Cancer and Stellate Cells. Cancers (Basel) 2020;12.
https://doi.org/10.3390/cancers12030640 |
| 82 | Ferdek PE, Jakubowska MA, Gerasimenko JV, Gerasimenko OV, Petersen OH: Bile acids induce necrosis in pancreatic stellate cells dependent on calcium entry and sodium‐driven bile uptake. The Journal of Physiology 2016;594:6147-6164.
https://doi.org/10.1113/JP272774 |
| 83 | Wang T, Wang Q, Pan G, Jia G, Li X, Wang C, Zhang L, Zuo C: ASIC1a Involves the Acid-Mediated Activation of Pancreatic Stellate Cells Associated With Autophagy Induction. Physiological Research 2023;72:49-57.
https://doi.org/10.33549/physiolres.934950 |
| 84 | Zhu L, Yin J, Zheng F, Ji L, Yu Y, Liu H: ASIC1 inhibition impairs the proliferation and migration of pancreatic stellate cells induced by pancreatic cancer cells. Neoplasma 2021;68:174-179.
https://doi.org/10.4149/neo_2020_200803N811 |
| 85 | Alvarez de la Rosa D, Zhang P, Shao D, White F, Canessa CM: Functional implications of the localization and activity of acid-sensitive channels in rat peripheral nervous system. Proc Natl Acad Sci U S A 2002;99:2326-2331.
https://doi.org/10.1073/pnas.042688199 |
| 86 | Petho Z, Najder K, Beel S, Fels B, Neumann I, Schimmelpfennig S, Sargin S, Wolters M, Grantins K, Wardelmann E, Mitkovski M, Oeckinghaus A, Schwab A: Acid-base homeostasis orchestrated by NHE1 defines the pancreatic stellate cell phenotype in pancreatic cancer. JCI Insight 2023;8.
https://doi.org/10.1172/jci.insight.170928 |
| 87 | Rotti PG, Xie W, Poudel A, Yi Y, Sun X, Tyler SR, Uc A, Norris AW, Hara M, Engelhardt JF, Gibson-Corley KN: Pancreatic and Islet Remodeling in Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Knockout Ferrets. The American Journal of Pathology 2018;188:876-890.
https://doi.org/10.1016/j.ajpath.2017.12.015 |
| 88 | Witt H, Apte MV, Keim V, Wilson JS: Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology 2007;132:1557-1573.
https://doi.org/10.1053/j.gastro.2007.03.001 |
| 89 | Leir SH, Tkachenko S, Paranjapye A, Meckler F, Van Wettere AJ, Kerschner JL, Kuznetsov E, Schacht M, Gillurkar P, Regouski M, Viotti Perisse I, Marriott CM, Liu Y, Bunderson I, White KL, Polejaeva IA, Harris A: Stellate cells are in utero markers of pancreatic disease in cystic fibrosis. Mol Med 2024;30:115.
https://doi.org/10.1186/s10020-024-00871-2 |
| 90 | den Hartog GJ, Qi S, van Tilburg JH, Koek GH, Bast A: Superoxide anion radicals activate hepatic stellate cells after entry through chloride channels: a new target in liver fibrosis. Eur J Pharmacol 2014;724:140-144.
https://doi.org/10.1016/j.ejphar.2013.12.033 |
| 91 | Dibaba D, Xun P, Yokota K, White E, He K: Magnesium intake and incidence of pancreatic cancer: the VITamins and Lifestyle study. British journal of cancer 2015;113:1615-1621.
https://doi.org/10.1038/bjc.2015.382 |
| 92 | Byeon S, du Toit-Thompson T, Hipperson L, Maloney S, Wenzel R, Gill AJ, Samra JS, Mittal A, Sahni S: Serum and tissue metallome of pancreatic ductal adenocarcinoma. Cancer Sci 2024;115:1446-1458.
https://doi.org/10.1111/cas.16124 |
| 93 | Nasulewicz A, Wietrzyk J, Wolf FI, Dzimira S, Madej J, Maier JA, Rayssiguier Y, Mazur A, Opolski A: Magnesium deficiency inhibits primary tumor growth but favors metastasis in mice. Biochimica et biophysica acta 2004;1739:26-32.
https://doi.org/10.1016/j.bbadis.2004.08.003 |
| 94 | Vaziri-Gohar A, Cassel J, Mohammed FS, Zarei M, Hue JJ, Hajihassani O, Graor HJ, Srikanth YVV, Karim SA, Abbas A, Prendergast E, Chen V, Katayama ES, Dukleska K, Khokhar I, Andren A, Zhang L, Wu C, Erokwu B, Flask CA, Zarei M, Wang R, Rothermel LD, Romani AMP, Bowers J, Getts R, Tatsuoka C, Morton JP, Bederman I, Brunengraber H, Lyssiotis CA, Salvino JM, Brody JR, Winter JM: Limited nutrient availability in the tumor microenvironment renders pancreatic tumors sensitive to allosteric IDH1 inhibitors. Nat Cancer 2022;3:852-865.
https://doi.org/10.1038/s43018-022-00393-y |
| 95 | de Baaij JH, Hoenderop JG, Bindels RJ: Magnesium in man: implications for health and disease. Physiol Rev 2015;95:1-46.
https://doi.org/10.1152/physrev.00012.2014 |
| 96 | Navarro Silvera SA, Rohan TE: Trace elements and cancer risk: a review of the epidemiologic evidence. Cancer Causes Control 2007;18:7-27.
https://doi.org/10.1007/s10552-006-0057-z |
| 97 | Dolensek J, Rupnik MS, Stozer A: Structural similarities and differences between the human and the mouse pancreas. Islets 2015;7:e1024405.
https://doi.org/10.1080/19382014.2015.1024405 |
| 98 | Sunami Y, Haussler J, Kleeff J: Cellular Heterogeneity of Pancreatic Stellate Cells, Mesenchymal Stem Cells, and Cancer-Associated Fibroblasts in Pancreatic Cancer. Cancers (Basel) 2020;12.
https://doi.org/10.3390/cancers12123770 |
| 99 | Neuzillet C, Tijeras-Raballand A, Ragulan C, Cros J, Patil Y, Martinet M, Erkan M, Kleeff J, Wilson J, Apte M, Tosolini M, Wilson AS, Delvecchio FR, Bousquet C, Paradis V, Hammel P, Sadanandam A, Kocher HM: Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J Pathol 2019;248:51-65.
https://doi.org/10.1002/path.5224 |
| 100 | Zhang Z, Zhang H, Liu T, Chen T, Wang D, Tang D: Heterogeneous Pancreatic Stellate Cells Are Powerful Contributors to the Malignant Progression of Pancreatic Cancer. Front Cell Dev Biol 2021;9:783617.
https://doi.org/10.3389/fcell.2021.783617 |