The P53N236S Mutation Plays a Regulatory Role in Osteosarcoma Metastasis Via the Cholesterol-Hedgehog Pathway
bSchool of Medicine, Kunming University, Kunming, Yunnan Province, China, 650500
Keywords
Abstract
Background/Aims:
Osteosarcoma is the most common primary bone cancer affecting children and adolescents worldwide. Although many treatments for osteosarcoma have been explored, the overall survival rate for patients with metastatic osteosarcoma is only 20% due to the lack of understanding of the biological mechanisms by which osteosarcoma metastasis occurs. Therefore, it is important to uncover the molecular mechanism of metastasis in osteosarcoma.Methods:
We compared the migration ability of primary osteosarcoma cells from p53 knockout (p53null) and p53N236S knock-in (p53S) mice. Furthermore, via RNA-sequencing (RNA-seq) data from mouse embryonic fibroblast (MEF) cells, we explored the mechanism by which p53S regulates the cholesterol synthesis pathway and the Hedgehog signaling pathway in primary osteosarcoma cells.Results:
We found that the migration ability of primary tumor cells from p53S mice was increased both in vivo and in vitro . Further investigations revealed that the cholesterol content in p53S sarcoma tissue and primary cells was increased following the upregulation of 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). We subsequently observed that elevated cholesterol levels can regulate the Hedgehog (HH) pathway and lead to tumor metastasis. We subsequently treated p53S sarcoma cells with the cholesterol neutralizer methyl-β-cyclodextrin (MβCD) and an HH pathway inhibitor; consequently, we reported that total cholesterol levels reduced both Hedgehog pathway activity and cell migration, whereas HH pathway activity reduced only cell migration.Conclusion:
In summary, we confirmed the enhanced metastatic ability of p53S sarcoma primary cells via in vivo and in vitro experiments and preliminarily confirmed the mechanism by which p53S promotes cholesterol synthesis and further activates the HH signaling pathway, thus leading to sarcoma metastasis. This study provides a theoretical basis for further revealing the function and mode of action of p53 mutations in the process of sarcoma metastasis, thereby providing a new potential target for the targeted diagnosis and treatment of sarcoma.Introduction
Osteosarcoma is the most prevalent primary bone malignancy affecting children and adolescents worldwide. Patients with metastatic osteosarcoma can experience an especially poor prognosis [1]. The main treatment methods for osteosarcoma involve surgery combined with chemotherapy and/or radiotherapy [2]. Although researchers have explored many therapeutic methods for osteosarcoma, the overall survival rate of patients with metastatic osteosarcoma is only 20% due to the lack of understanding of the biological mechanism of metastasis. Therefore, the elucidation of the molecular mechanism of osteosarcoma metastasis is very important.
Gene mutations represent one of the main factors leading to the development of tumors. Moreover, p53 gene mutations are found in 50% of human tumors, and the p53 mutation rate in osteosarcoma patients has been demonstrated to be as high as 80%[3, 4]. Mutated p53 can promote the motility, invasion and metastasis of cancer cells while partially losing the function of wild-type p53[5]. However, reports concerning the relationship between p53 mutations and osteosarcoma metastasis, as well as the underlying mechanism, are scarce. Recent studies have reported that the p53R172H mutation (the human p53R175H mutation, also known as p53H) upregulates the expression of the immune-related gene ONZIN (Plac8) and further activates the CXCL5-MAPK signaling pathway to promote osteosarcoma metastasis [6].
We have previously reported the existence of p53N236S (p53N239S in humans, also known as p53S) in three independent tumorigenic mouse cell lines derived from telomere dysfunction. p53S has been reported as one of the less common but recurrent mutations in human cancers and has been labeled as a hotspot mutation in the TCGA database [7]. In our previous study, we generated p53S knock-in mice and reported that the sarcoma metastasis rate of p53S mutant mice was greater than that of p53null mice [8]. However, our RNA-seq data revealed that the expression of the Plac8 gene in tumors from p53S mice was significantly lower than that in tumors from p53null mice, thus suggesting that the mechanism by which p53S leads to increased osteosarcoma metastasis may differ from that associated with p53H mutation.
Studies have shown that abnormal cholesterol synthesis and transport can affect the structure of the phospholipid bilayer, thereby affecting the fluidity of cellular biomembranes and consequently affecting the metastatic ability of tumor cells; moreover, lipids can affect the course of disease by remodeling mitochondrial proteins [9]. The Hedgehog (Hh) protein is the first endomorph modified by cholesterol and plays a key role in development and tumorigenesis [10]. The Hh protein precursor is broken down into two parts, including the carboxyl domain (HhC) and the amino domain (HhN), which are released into the extracellular fluid via cholesterol modification and activate the Hh pathway, thus promoting tumor development and metastasis.
This study aims to elucidate the mechanism by which p53S enhances osteosarcoma metastasis via a comparative analysis of various genetic variants of p53 in primary osteosarcoma cell models. This study provides foundational insights into how p53S mutations modulate the cholesterol-Hh pathway, thereby increasing the metastatic potential of osteosarcoma; moreover, the results contribute to a deeper understanding of the role that mutated p53 plays in tumor metastasis.
Materials and Methods
Cell culture
The mouse embryonic fibroblast (MEF) cell lines that were used for RNA-seq were cultured in DMEM supplemented with 10% fetal bovine serum (HyClone, CA), along with 100 units/mL penicillin and 100 μg/mL streptomycin sulfate, in a 3% oxygen and 5% CO2 incubator at 37 °C. The primary tumor cell lines were cultured in DMEM supplemented with 10% fetal bovine serum (HyClone, CA), along with 100 units/mL penicillin and 100 μg/mL streptomycin sulfate, in a 5% CO2 incubator at 37 °C. MβCD (5%, Solarbio) and vismodegib [11] (40 μM, Beyotime) were incubated with the cells to explore the involved mechanisms.
Animal experiments
For the mouse tail vein injections, male mice were placed in a fixator with their tails exposed, and the cells were injected into the tail vein at a dose of 0.2 mL/mouse (0.5×10^6 cells/mL). The two groups of mice were sacrificed after the experiment, and lung, liver, spleen and kidney tissues were collected for imaging.
Detection of total cholesterol (enzymatic hydrolysis method)
The cells were normally grown in high-glucose DMEM containing 10% fetal bovine serum. For the cell experiments, cells in the logarithmic growth phase were collected, and the cell density was adjusted to 1×10^5/mL. Subsequently, 200 μL of RIPA buffer was added to the cells in each well, followed by centrifugation of the supernatant. A portion of the supernatant was utilized for the determination of the intracellular TC level, and another portion was used for protein extraction. Total cholesterol and total protein concentrations were measured according to the instructions of the tissue total cholesterol kit (Beyotime) and the instructions of the BCA protein concentration kit, respectively. Finally, the OD data were measured with a synergy/H1 microplate reader (BioTek), and the results are expressed as the relative levels of cholesterol per mg of protein.
Real-time fluorescence quantitative PCR (qRT-PCR)
For qRT-PCR analysis, total RNA was extracted using TRIzol reagent (Invitrogen), and 1 μg of total RNA was converted to cDNA via a high-capacity cDNA reverse transcription kit (4368813; Applied Biosystems, Waltham, MA, USA). Moreover, qPCR was performed via 45 cycles of iQ SYBR Green Supermix (1708880; Bio-Rad, Hercules, CA, USA) with a CFX Connect Real-time system (Bio-Rad), and the fold change for all of the samples was calculated via the 2−∆∆Ct method. Peptidylprolyl isomerase A (PPIA) was used as an internal reference. The utilized primers were as follows: Srebf2 (forward primer: 5› CAGGTGCAGACGGTACAGG 3›, reverse primer: 5› CGACCCTTACTGGCACTTGAA 3›), Hmgcr (forward primer: 5› CTTTCAGAAACGAACTGTAGCTCAC 3›, reverse primer: 5› CTAGTGGAAGATGAATGGACATGAT 3›), Shh (forward primer: 5› AAAGCTGACCCCTTTAGCCTA 3›, reverse primer: 5› GTCGGAGTTTCTTGTGATCTTC 3›), Smo (forward primer: 5› CCCTGCTGTGTGCTGTCTAC 3›, reverse primer: 5› GTGTGCAACGCAGAAAGTCAG 3›), Ptch1 (forward primer: 5› AAAGAACTGCGGCAAGTTTTTG 3›, reverse primer: 5› CTTCTCCTATCTTCTGACGGGT 3›), Gli1 (forward primer: 5› TGGAAGGGGACATGTCTAGC 3›, reverse primer: 5› GCTCACTGTTGATGTGGTGC 3›), Gli2 (forward primer: 5› CAGTGGCAGTTGGTCTCGT 3›, reverse primer: 5› ATAAGCGGAGCAAGGTCAAG 3›), Gli3 (forward primer: 5› CCCTCTCTCCCTTATCGTG 3›, reverse primer: 5› AAGGCAAGTCTGGATACGTT 3›), and Gapdh (forward primer: 5› GTCTTCACCACCATGGAGAAGGC 3›, reverse primer: 5› TTGTTGTCATGGATGACCTTGGCC 3›).
Transwell assay
Transwell assays are one method for detecting cell migration ability. The upper compartment is designated as the upper chamber, whereas the culture plate is referred to as the lower chamber. The upper chamber holds the upper culture medium, and the lower chamber contains the lower culture fluid. The upper and lower culture fluids are separated by a polycarbonate membrane. The filter pore size of the polycarbonate membrane is 8 μm, and the cells can pass through these pores. The cells were seeded in the upper chamber. Due to the permeability of the polycarbonate membrane, the components in the lower culture medium can affect the number of cells in the upper chamber; thus, the migration ability of the cells can be analyzed.
Scratch test
The cell scratch assay is a simple, inexpensive method and one of the first methods that was developed to assess directed cell migration in vitro ; moreover, this method mimics the process of cell migration during in vivo healing. The effects of formononetin on the adhesive and invasive abilities of sarcoma cells derived from different genotypes were detected with a scratch test. First, the cell suspension was added to the plates at a concentration of approximately 1×10^5 cells/well and cultured with 5% CO2 at 37 ℃ overnight. Afterwards, transverse lines perpendicular to the straight lines were drawn on the backs of these plates. Subsequently, after washing with PBS, the cells were incubated for 24 h. Finally, cell migration was observed and photographed with an optical microscope.
Western blot
The expression of cholesterol synthesis- and HH pathway-related proteins was verified by using Western blot analysis.
The cell samples were lysed in RIPA buffer containing protease inhibitor. A total of 15 μg of total protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a polyvinylidene fluoride (PVDF) membrane. After being blocked in 5% nonfat milk for 1 hour at room temperature, the membranes were incubated with primary antibodies (either overnight at 4 ℃ or for 2 hours at room temperature). The membranes were then incubated with horseradish peroxidase-labeled secondary antibodies and visualized with enhanced chemiluminescence (ECL).
The following primary antibodies were used: anti-p53 (Santa Cruz), anti-SREBP2 (Abcam), anti-HMGCR (ABclonal), anti-Gli1 (ABclonal), anti-SHH (ABconal), anti-GAPDH (Abcam), and anti-α-tubulin (Millipore).
Filipin staining
The cells that were grown on glass cover slips were rinsed three times with PBS and subsequently fixed for 10 minutes at room temperature with 3.7% formaldehyde in PBS. The cells were then stained for 2 hours at room temperature in the dark with 50 μg/mL filipin (Sigma). Finally, the cells were rinsed three times (with each rinse occurring for 5 minutes) with PBS. The cells were subsequently mounted on slides. Images were then acquired by a fluorescence microscope (Nikon, Japan).
RNA-seq and gene set enrichment assay (GSEA)
The cell samples were collected and sent for commercial RNA sequencing services (Novogene, China). To calculate the fold changes, we used R version 3.5.1 (Institute for Statistics and Mathematics, Vienna, Austria; https://www.r-project.org) to perform edge R analysis and to normalize the RNA-seq reads. The normalized RNA-seq reads were also subjected to GSEA v4.1.0 for Windows (Massachusetts Institute of Technology, and Regents of the University of California; http://www.gsea-msigdb.org/gsea/downloads.jsp) to perform a gene set enrichment assay (GSEA).
Statistical analysis
Immunoblotting quantification and grayscale analysis were performed using ImageJ software. Statistical analysis and graph preparations were performed using GraphPad Prism 8. The measurement data are expressed as the means ± SDs, and group comparisons were performed using a t test, ANOVA test or log-rank (Mantel-Cox) test. A p value < 0.05 was deemed to be statistically significant.
Results
The migration ability of p53S tumor cells is greater than that of p53null cells both in vitro and in vivo
To elucidate the differential migration capabilities of osteosarcoma cells with distinct p53 genetic backgrounds, we conducted a comparative analysis of p53null and p53S genotypes(Fig 1A) utilizing both scratch assays and Transwell migration assays in vitro . The results demonstrated markedly enhanced closure of the scratch wounds by p53S osteosarcoma cells at 24 hours post injury, as illustrated in Fig. 1B. Furthermore, Transwell assays revealed that, compared to p53null cells, a significantly greater number of p53S cells translocated through the membrane at 12 hours after seeding, as depicted in Fig. 1C. These findings unequivocally suggest a superior migratory capacity of p53S osteosarcoma cells over p53null cells in vitro .
After expanding our investigation to in vivo models, we assessed the metastatic propensity of these genetically distinct sarcoma cells. Using 8-week-old C57BL/6 mice as hosts, we intravenously administered 0.5×10^6 cells of each genotype and monitored subsequent tumor progression, weight fluctuations, and survival, which resulted in the generation of weight (Fig. 2B) and survival curves (Fig. 2C). Notably, mice inoculated with p53S cells began to succumb to the tumor burden at Day 12 postinoculation, thus necessitating euthanasia. Examinations of the pulmonary tissue from these mice revealed dense aggregations of nodules in the lungs of mice injected with p53S cells, which was markedly different from the smoother lung surfaces observed in p53null cell-injected mice, as shown in Fig. 2D. Quantitative analyses further revealed significant increases in both the lung index and wet weight ratio in p53S-injected mice (Fig. 2E & F), with hematoxylin and eosin staining revealing a greater prevalence of cancerous nodules in lungs harboring p53S cells compared to those harboring p53null cells (Fig. 2J); moreover, there were no discernible impacts observed on other organs (Fig. 2G-J). Additionally, we counted the number of nodules in the HE-stained lung sections and found that the number of lung nodules in mice inoculated with p53S tumor cells was significantly higher than that in mice inoculated with p53null tumor cells (Fig. 2K). Collectively, these in vivo results corroborate the enhanced metastatic capabilities of p53S sarcoma cells, thereby underscoring the pivotal role of p53 stabilization in modulating the metastatic behavior of osteosarcoma cells.
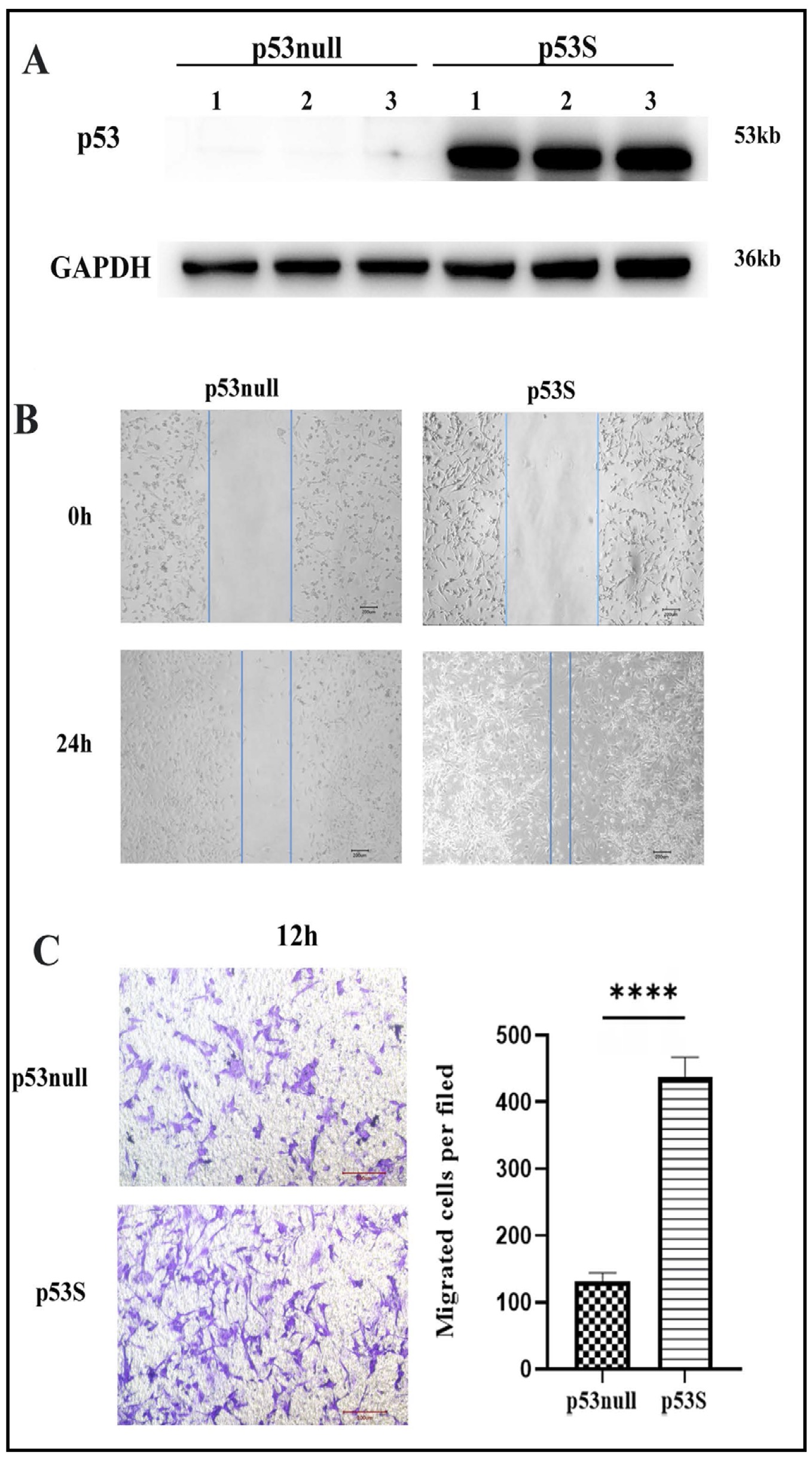
Fig. 1: The migration ability of p53S tumor cells is greater than that of p53null tumor cells in vitro. A. Expressions of p53 via the WB experiment. B. Images of the wounds of the cell layer at 0 h and after 24 h showing the widths of the scratches with different p53 phenotypes on the migratory ability. C. Images and cell counts from the migration assay performed via the transwell assay at 12 h. ***p<0.005.
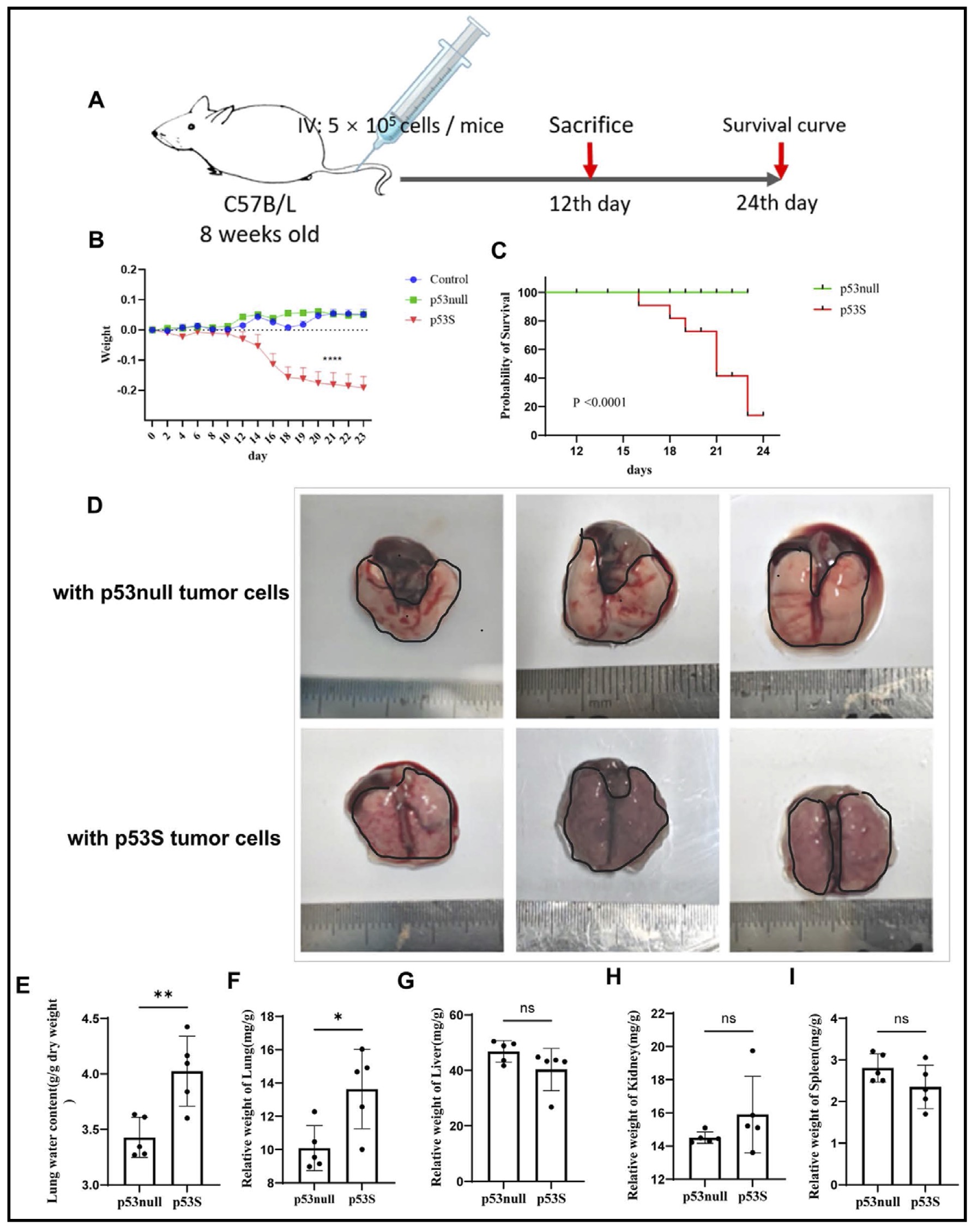
Fig. 2: The migration ability of p53S tumor cells is greater than that of p53null tumor cells in vivo. A. Experimental plan. B. Changes in the weights of the tumor-bearing mice. C. Survival curves of tumor-bearing mice. D. Lungs of mice bearing tumors for 12 days. E-I. Organ coefficients of tumor-bearing mice at 12 days. J. The lungs, livers, spleens and kidneys of tumor-bearing mice were stained with HE and observed with a 20 × microscope. K. Statistical chart of the quantity of tumor masses in HE-stained lung sections from tumor-bearing mice. ns, not significant; *p<0.05, **p<0.01, ***p<0.005, ****p<0.001.
The cholesterol content of p53S sarcoma cells is greater than that of p53null sarcoma cells
Emerging research suggests that cholesterol homeostasis plays a critical role in tumor metastasis. Our RNA-seq data demonstrated the activation of the cholesterol synthesis pathway in p53S MEF cells (Fig. 3A). We subsequently observed a substantial increase in total cholesterol levels in both p53S sarcoma tissues and primary cells, as documented in Fig. 3B and C. Via filipin staining, we further demonstrated a significant increase in cholesterol levels in p53S cells (Fig. 3D).
In-depth analysis of our transcriptome data allowed us to explore cholesterol metabolism in sarcoma cells with different p53 genetic backgrounds. Notably, p53S sarcoma cells exhibited a marked increase in the expression of HMGCR, which is the rate-limiting enzyme involved in cholesterol synthesis, thus indicating that the accumulation of cholesterol in these cells is primarily due to increased synthesis (Fig. 3E-J). Given that SREBP2 is the transcription partner of HMGCR and that p53H transcriptionally upregulates SREBP2, we examined the expression of SREBP2 by WB and RT-qPCR. Interestingly, we did not observe a significant change in SREBP2 in p53S tumor cells. These results indicated that p53S promoted cholesterol accumulation via the direct targeting of HMGCR.
To ascertain whether the increased cholesterol content directly contributes to the increased metastatic potential of p53S cells, we treated these cells with the cholesterol-sequestering agent methyl-β-cyclodextrin (MβCD). Treatment with MβCD resulted in a significant reduction in migratory capacity, as illustrated in Fig. 3K and L. These findings support the hypothesis that elevated cholesterol levels are pivotal in facilitating the enhanced metastatic ability of p53S sarcoma cells.
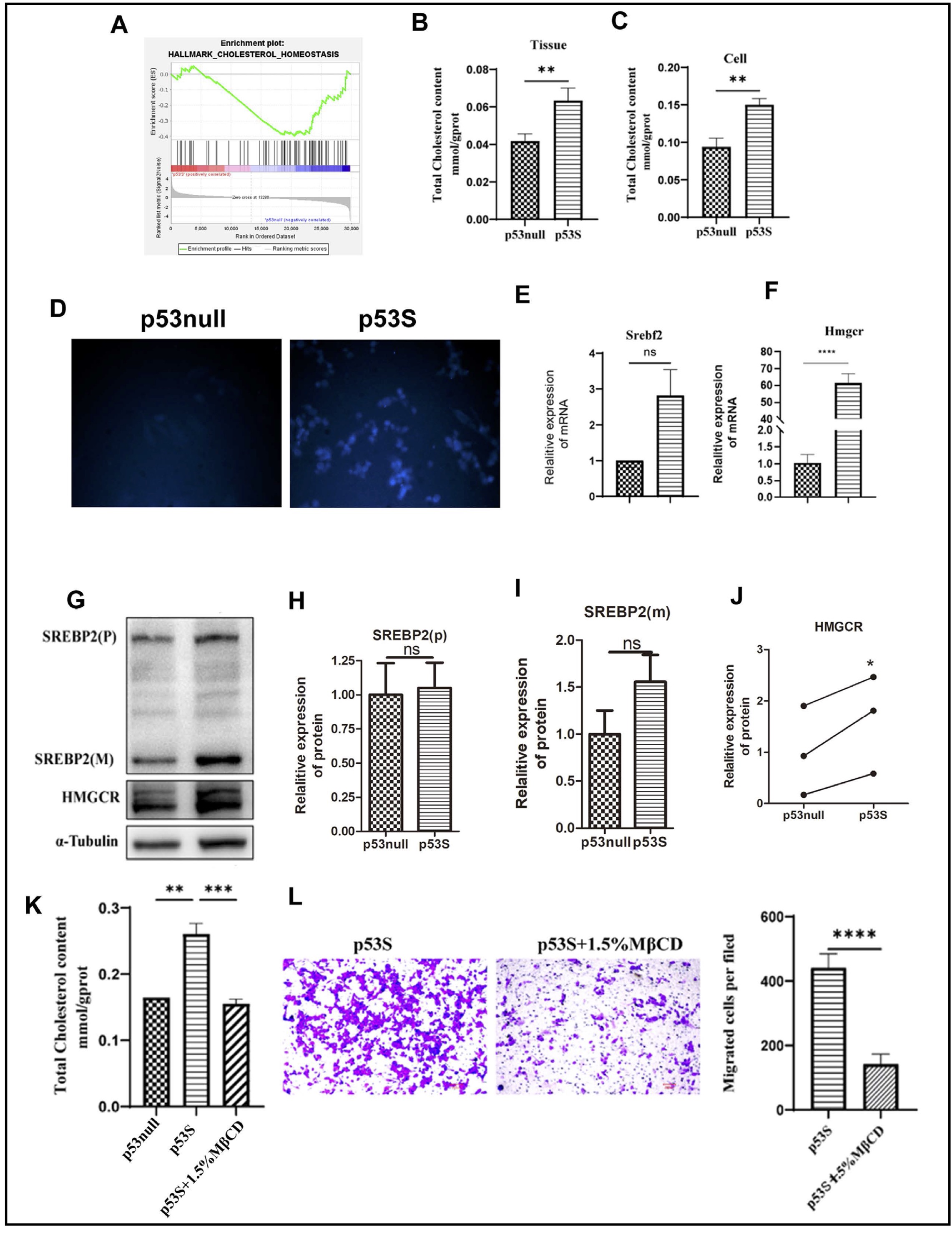
Fig. 3: Mutant p53S promotes cholesterol synthesis by promoting the expression of HMGCR (a rate-limiting enzyme) and leading to tumor metastasis. A. GSEA of the cholesterol homeostasis gene set. B-C. The cholesterol content in p53null and p53S osteosarcoma tissues and cells was measured by enzymatic hydrolysis. D. The fluorescence intensity of cholesterol was measured via the filipin test, and observations were made using a fluorescence microscopy. E. The relative mRNA expression of Srebf2. F. The relative mRNA expression of Hmgcr. G-J. Protein expression of SREBP2 and HMGCR. K. Cholesterol content in primary sarcoma cells treated with MβCD. L. Photographs and cell counts of the migration assay conducted by the transwell assay after MβCD treatment. ns, not significant; *p<0.05, **p<0.01, ***p<0.005, ****p<0.001.
The Hedgehog pathway is activated in p53S sarcoma cells
Based on our previous findings that the augmented migratory capacity of p53S sarcoma cells is associated with increased intracellular cholesterol accumulation, we sought to elucidate the mechanistic link between cholesterol and cellular migration. Cholesterol can directly modulate key ligands, such as Sonic Hedgehog (SHH) and Smoothened (SMO) proteins, of the Hedgehog (HH) signaling pathway, thereby facilitating its activation (a process that is integral to tumor metastasis). In alignment with these findings, our transcriptomic analysis demonstrated the activation of the HH pathway in p53S cells (Fig. 4A).
To further investigate this phenomenon, we compared Hedgehog pathway activity across sarcoma cells with differing p53 statuses. Our findings revealed no significant differences in the transcription levels of the pathway ligand gene Shh, the signal transducer gene Smo or the pathway inhibition marker Gli3 between p53S and p53null sarcoma cells. However, notable upregulation of the transcriptional levels of the effector protein-encoding genes Gli1 and Gli2 was detected in p53S cells, as depicted in Fig. 4B. Additionally, the concentration of N-SHH (a pathway ligand) was found to be elevated in the culture supernatant of p53S cells, as shown in Fig. 4C. Concurrently, Gli1 protein expression, which is a widely recognized marker of pathway activation, was increased in p53S osteosarcoma cells (Fig. 4D and E), thus suggesting the activation of the HH signaling pathway in these cells.
To ascertain whether the activation of the HH signaling pathway was driven by cholesterol accumulation, we treated p53S sarcoma cells with the cholesterol neutralizer MβCD. The post treatment results demonstrated that the neutralization of cholesterol effectively inhibited Hedgehog pathway activity, as shown in Fig. 4F and G. These findings support the hypothesis that cholesterol accumulation is a critical upstream regulator of Hedgehog pathway activation in p53S osteosarcoma cells, thereby could be contributing to their enhanced migratory and metastatic capacities.
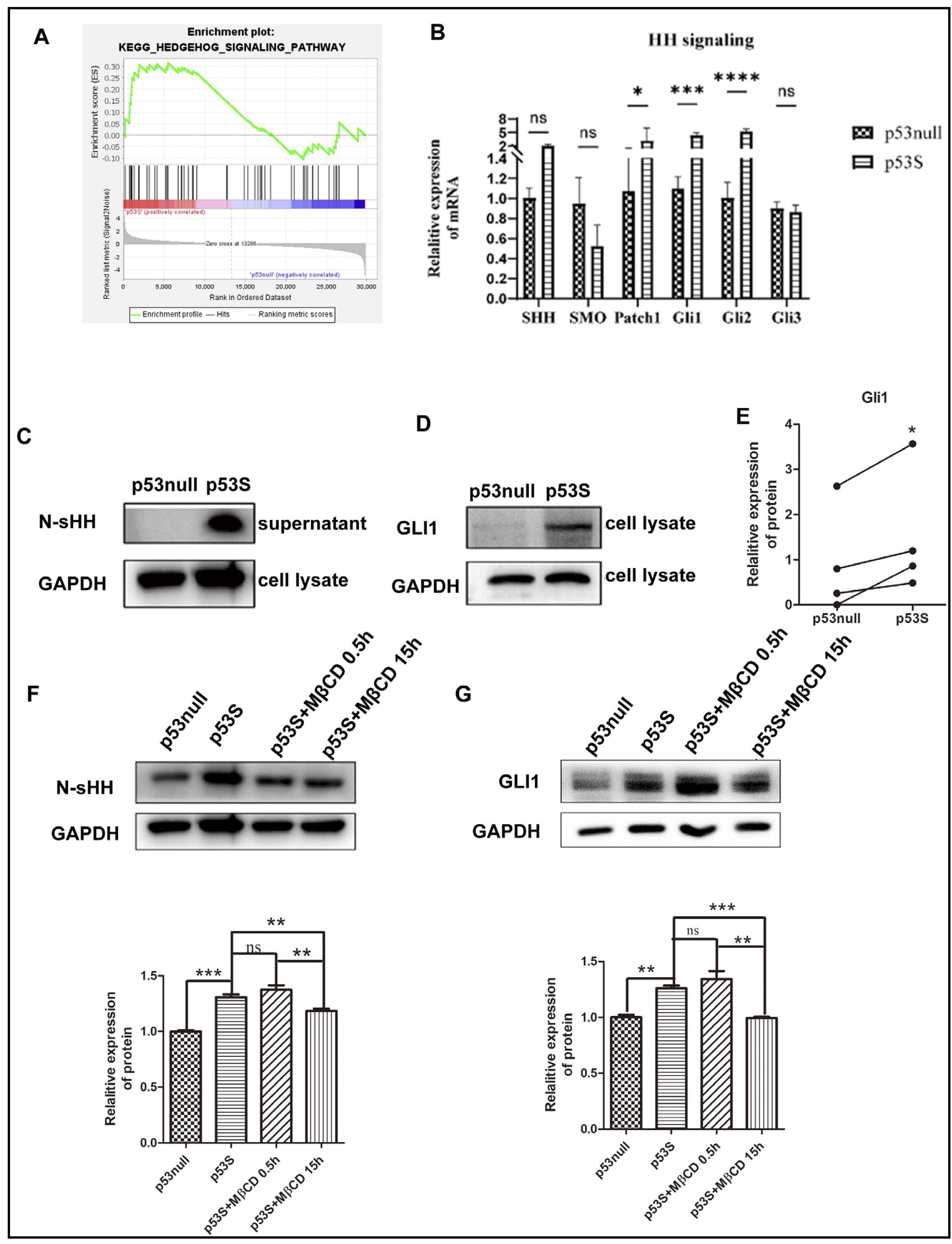
Fig. 4: Elevated p53S cholesterol activates the HH pathway. A. GSEA of the HH signaling pathway. B. mRNA expression levels of HH pathway-associated genes. C. The protein expression level of N-SHH in the cell culture supernatant. D-E. The protein expression level of Gli1. F. The protein expression level of N-SHH after MβCD treatment. G. The protein expression level of Gli1 after MβCD treatment. ns, not significant; *p<0.05, **p<0.01, ***p<0.005, ****p<0.001.
Inhibition of the Hedgehog pathway reduces the migration ability of p53S sarcoma cells
Research has shown that the activation of the HH signaling pathway plays a pivotal role in promoting tumor metastasis [12]. To substantiate the premise that the increased metastatic ability of p53S osteosarcoma cells is attributable to HH pathway activation, we administered vismodegib (40 μM), which is a known inhibitor of the HH signaling pathway [11], to p53S osteosarcoma cells. Upon the addition of the vismodegib, the expression levels of the signaling genes Smoothened (Smo) and Patched 1 (Ptch1) remained unchanged (Fig. 5A & B). However, the expression of the pathway-activating marker genes Gli1 and Gli2 was downregulated (Fig. 5C & D), while the expression of the pathway-inhibitory marker gene Gli3 was upregulated (Fig. 5E). Additionally, the expression levels of the active ligand N-Shh and the Gli1 protein were both suppressed (Fig. 5F-J). These results indicate that the Hedgehog (HH) pathway in p53S was inhibited by the inhibitor. Concomitantly, the migratory ability of vismodegib-treated p53S osteosarcoma cells was notably diminished, as depicted in Fig. 5K and L. Interestingly, this treatment did not affect the cholesterol levels within the cells (Fig. 5M). These findings collectively demonstrate that the increased metastatic potential of p53S osteosarcoma cells is mediated via the activation of the HH signaling pathway and that cholesterol accumulation occurs upstream of the HH signaling pathway.
Our study demonstrated that the p53S phenotype upregulated the expression of HMGCR, which is a key regulator of cholesterol biosynthesis. This upregulation activates cholesterol synthesis pathways, thus culminating in elevated cholesterol levels within sarcoma cells. The increased cholesterol content then facilitates osteosarcoma progression by activating the Hedgehog signaling pathway. These mechanistic insights are succinctly depicted in Fig. 6.
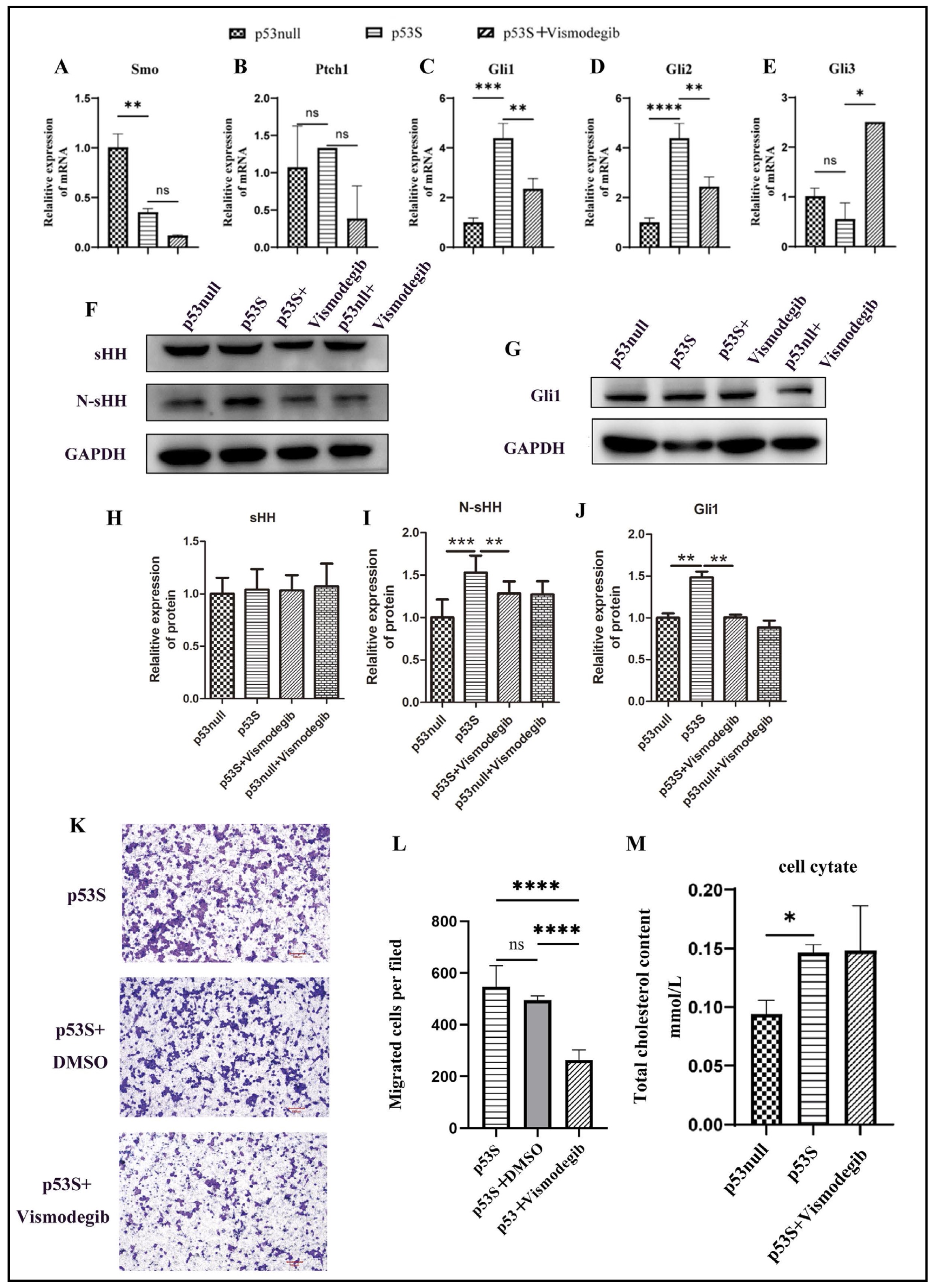
Fig. 5: Inhibition of the Hedgehog pathway reduces the migration ability of p53S sarcoma cells but does not affect cholesterol synthesis. A-E. mRNA expression levels of HH pathway-associated genes after vismodegib (40 μM) treatment. F-J. The protein expression levels of sHH, N-sHH and Gli1 after vismodegib (40 μM) treatment. K-L. Photographs and cell counts of the migration assay results from the transwell assay after vismodegib (40 μM) treatment. M. Cholesterol content in sarcoma cells after vismodegib treatment. ns, not significant; *p<0.05, **p<0.01, ***p<0.005, ****p<0.001.
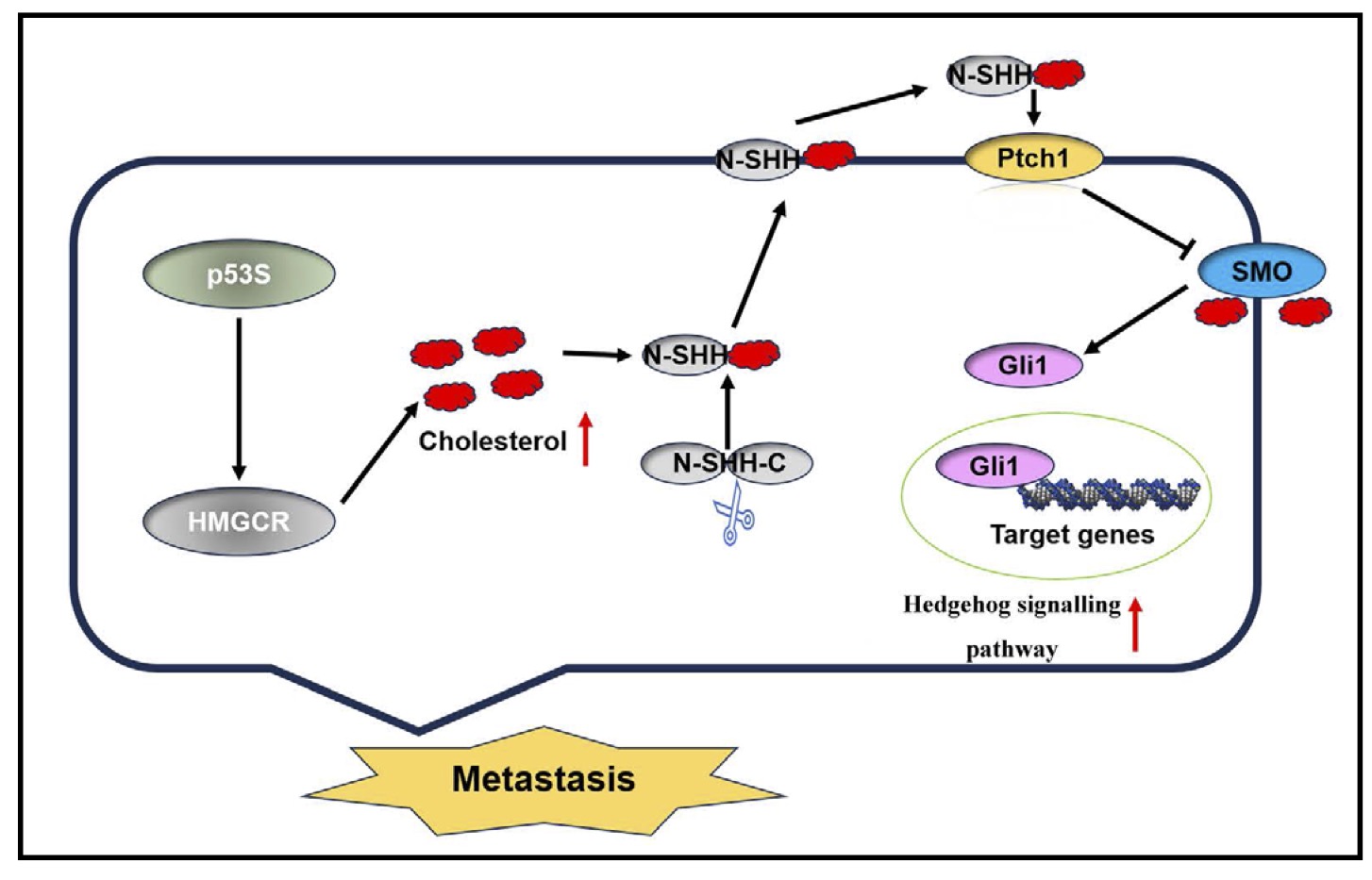
Fig. 6: p53S regulates the cholesterol-activated Hedgehog pathway to promote metastasis in sarcoma cells.
Discussion
Tumor metastasis is a predominant cause of mortality among cancer patients. Despite comprehensive research efforts, the mechanisms underlying tumor metastasis continue to pose a significant scientific challenge. Metastatic cancer cells demonstrate selectivity at each phase of the metastatic cascade, whereby they adjust their metabolism in a dynamic manner [13]. In particular, sarcomas exhibit pronounced malignancy in humans, which frequently leads to lung metastases and a correspondingly substantial reduction in patient survival [14].
Previous studies in our laboratory revealed that the number of primary tumors and their rate of metastasis in p53S mice were increased [8]. Another study revealed that p53S plays an important role in cancer-associated fibroblasts (CAFs), which serve important roles in cancer development and progression, via the activation of the Stat3 pathway [15]. Furthermore, in vivo assessments revealed that these cells exhibited pronounced lung metastasis, increased pulmonary edema, and significantly decreased survival rates when injected into the tail vein. These findings underscore the role of p53S in enhancing osteosarcoma metastasis. A previous study reported that p53H mutations can upregulate the expression of the immune-related gene ONZIN (Plac8) and further activate the CXCL5-MAPK signaling pathway to promote osteosarcoma metastasis [6]. However, our RNA-seq data revealed that the expression of the Plac8 gene in the tumors of p53S mice was significantly lower than that in the p53null group. These results indicate that p53S increases the metastasis of osteosarcoma via another mechanism.
Cholesterol homeostasis is critical for maintaining normal cell function, and an imbalance in cholesterol homeostasis often occurs during tumor development and progression. Multiple studies have shown that the upregulation of genes that are involved in fatty acid and cholesterol synthesis is an important factor in accelerating tumor progression; moreover, the dysfunction of the cholesterol synthesis pathway is associated with increased tumor malignancy. An increasing number of studies have shown that p53 regulates lipid metabolism via transcriptional control or protein-protein interactions. Furthermore, studies have shown that wild-type p53 can inhibit the cholesterol synthesis pathway, whereas mutant p53 promotes tumor metastasis by regulating the cholesterol synthesis pathway [16]. This study revealed that the content of lipid droplets in p53S tumors was increased, and we speculated that the cholesterol synthesis pathway may be activated to further promote cholesterol synthesis. We confirmed that the mRNA and protein expression of HMGCR was increased and that the intracellular cholesterol content was also increased, thus suggesting that p53S may promote HMGCR to further promote cholesterol synthesis. This process is different from the function of p53H in regulating cholesterol synthesis [16]. However, the mechanism by which p53S promotes the expression of HMGCR remains to be further investigated.
Previous studies have shown that abnormal cholesterol synthesis and transport can affect the structure of the phospholipid bilayer, thereby affecting the fluidity of cellular biomembranes and consequently affecting the metastatic ability of tumor cells; moreover, lipids can affect the course of disease by remodeling mitochondrial proteins [9]. Additionally, studies have reported that overloaded cholesterol content enhances the EMT pathway, which subsequently promotes tumor development [17]. Cholesterol and its derivatives can activate the HH pathway by promoting the release of HH pathway ligands and can directly act on the key transduction factor SMO of this pathway [18]. Moreover, 70% of OS samples are estimated to demonstrate aberrant expression of components of the HH signaling pathway [19-21]. In conjunction with the GSEA of the mRNA-seq data, we found that the HH pathway activity of p53S was increased. Furthermore, we treated p53S osteosarcoma cells with the cholesterol neutralizer MβCD and found that the HH pathway was inhibited. These findings indicate that p53S may indirectly activate the HH pathway by promoting cholesterol synthesis. However, some studies have reported that the activation of the HH pathway can promote the synthesis of cholesterol; thus, in p53S tumor cells, it is unknown as to whether the increase in cholesterol synthesis and the activation of the HH pathway represents a bidirectional effect. To address this issue, we treated p53S cells with an HH pathway inhibitor, analyzed the cholesterol synthesis pathway and cholesterol content, and found that the inhibition of the HH pathway did not affect cholesterol synthesis. These findings indicate that p53S promotes cholesterol synthesis, which is an important mechanism of activating the HH signaling pathway. These results suggest that in p53S osteosarcoma cells, elevated cholesterol promotes the secretion of the SHH protein; however, whether it directly binds to SMO to activate the HH pathway warrants further investigations.
In conclusion, we confirmed the enhanced metastatic ability of p53S sarcoma primary cells via in vivo and in vitro experiments and preliminarily confirmed the mechanism by which p53S promotes cholesterol synthesis and further activates the HH signaling pathway, thus leading to sarcoma metastasis. This study provides a theoretical basis for further revealing the function and mode of action of p53 mutations in the process of sarcoma metastasis, thereby providing a new potential target for the targeted diagnosis and treatment of sarcoma.
Limitations and Future Directions
We recognize that our study is largely correlative and does not provide direct mechanistic evidence for the observed relationships between p53S, cholesterol biosynthesis, and Hedgehog pathway activation. Techniques such as chromatin immunoprecipitation (ChIP), co-immunoprecipitation, or reporter assays would strengthen the claim that p53S directly regulates HMGCR. Additionally, the modest changes observed in Hedgehog pathway activity markers warrant further investigation to confirm their biological relevance. While we employed single doses for vismodegib and methyl-β-cyclodextrin treatments based on prior studies, dose-response experiments and side-by-side comparisons in p53S and p53null cells would provide a more comprehensive understanding. Despite these limitations, this study establishes a foundational link between p53S and osteosarcoma metastasis and identifies cholesterol metabolism and Hedgehog signaling as potential therapeutic targets. In the subsequent research, we will conduct an in-depth exploration of the mechanism by which p53S upregulates HMGCR.
Acknowledgements
We thank Dr. Ying Luo from Guizhou Medical University for helpful suggestions regarding this study.
Author Contributions
Dan Juhua designed the study. Wang Lulin and Zhang Shuojie performed the experiments. Wang Lulin wrote the first draft of the manuscript. Wang Hui analyzed the RNA-seq data. Liu Jiawei provided a summary illustration, was responsible for organizing materials for submissions, and performed the additional experiments during revising. Dan Juhua, Xie Xiaoli and Jia Jing revised the manuscript. All of the authors read and approved the final manuscript.
Funding Sources
Contract grant sponsor: Applied Basic Research Foundation of Yunnan Province (202201AT070194; 202201AT070289); Joint Funds of the Science Foundation of Kunming University of Science and Technology and The First Peoples Hospital of Yunnan Province (KUST-KH2022009Y); Joint Funds of the Science Foundation of Kunming University of Science and Technology and The Peoples Hospital of Lijiang (KUST-LJ2022003Y); The Yunnan Applied Basic Research Program University Joint General Fund Project (202001BA070001–170).
Statement of Ethics
This study conforms to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23, revised in 1996 for scientific purposes).
The Ethics Committee of Kunming University of Science and Technology also approved this study (No. SYXK (Dian) K2018-0008).
Disclosure Statement
Not applicable.References
| 1 | Mirabello L, Troisi RJ, Savage SA: Osteosarcoma Incidence and Survival Rates From 1973 to 2004 Data From the Surveillance, Epidemiology, and End Results Program. Cancer-Am Cancer Soc 2009;115:1531-1543.
https://doi.org/10.1002/cncr.24121 |
| 2 | Meltzer PS, Helman LJ: New Horizons in the Treatment of Osteosarcoma. New Engl J Med 2021;385:2066-2076.
https://doi.org/10.1056/NEJMra2103423 |
| 3 | Babatunde O, Notti RQ, Tap WD: Characterizing TP53 mutations in bone and soft tissue sarcoma. J Clin Oncol 2024;42
https://doi.org/10.1200/JCO.2024.42.16_suppl.e23521 |
| 4 | Thoenen E, Curl A, Iwakuma T: TP53 in bone and soft tissue sarcomas. Pharmacol Therapeut 2019;202:149-164.
https://doi.org/10.1016/j.pharmthera.2019.06.010 |
| 5 | Tang QS, Su ZY, Gu W, Rustgi AK: Mutant p53 on the Path to Metastasis. Trends Cancer 2020;6:62-73.
https://doi.org/10.1016/j.trecan.2019.11.004 |
| 6 | Zhang YQ, Hu QH, Li GX, Li LL, Liang SL, Zhang Y, Liu JY, Fan ZF, Li L, Zhou BZ, Ruan YX, Yang XL, Chen SA, Mu TY, Wang GW, Xiong SB: ONZIN Upregulation by Mutant p53 Contributes to Osteosarcoma Metastasis Through the CXCL5-MAPK Signaling Pathway. Cell Physiol Biochem 2018;48:1099-1111.
https://doi.org/10.1159/000491976 |
| 7 | Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, Gao JJ, Socci ND, Solit DB, Olshen AB, Schultz N, Taylor BS: Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol 2016;34:155-+.
https://doi.org/10.1038/nbt.3391 |
| 8 | Zhao LJ, Wang BY, Zhao XL, Wu XM, Zhang QS, Wei CY, Shi ML, Li YL, Tang WR, Zhang JH, Yang JL, Singh SK, Jia ST, Luo Y: Gain of function in the mouse model of a recurrent mutation p53 promotes the formation of double minute chromosomes and the oncogenic potential of p19. Mol Carcinogen 2018;57:147-158.
https://doi.org/10.1002/mc.22737 |
| 9 | Liu CY, Chen H, Hu BC, Shi JJ, Chen YC, Huang K: New insights into the therapeutic potentials of statins in cancer. Front Pharmacol 2023;14
https://doi.org/10.3389/fphar.2023.1188926 |
| 10 | Mann RK, Beachy PA: Novel lipid modifications of secreted protein signals. Annu Rev Biochem 2004;73:891-923.
https://doi.org/10.1146/annurev.biochem.73.011303.073933 |
| 11 | Hehlgans S, Booms P, Güllülü Ö, Sader R, Rödel C, Balermpas P, Rödel F, Ghanaati S: Radiation Sensitization of Basal Cell and Head and Neck Squamous Cell Carcinoma by the Hedgehog Pathway Inhibitor Vismodegib. Int J Mol Sci 2018;19
https://doi.org/10.3390/ijms19092485 |
| 12 | Ram Kumar RM, Fuchs B: Hedgehog Signaling Inhibitors as Anti-Cancer Agents in Osteosarcoma. Cancers 2015;7:784-794.
https://doi.org/10.3390/cancers7020784 |
| 13 | Clevenger AJ, McFarlin MK, Gorley JPM, Solberg SC, Madyastha AK, Raghavan SA: Advances in cancer mechanobiology: Metastasis, mechanics, and materials. Apl Bioeng 2024;8
https://doi.org/10.1063/5.0186042 |
| 14 | Tan LY, Wang YT, Hu X, Min L: The Roles of Exosomes in Metastasis of Sarcoma: From Biomarkers to Therapeutic Targets. Biomolecules 2023;13
https://doi.org/10.3390/biom13030456 |
| 15 | Liu Q, Yu B, Tian YB, Dan JH, Luo Y, Wu XM: P53 Mutant p53 Regulates Cancer-Associated Fibroblasts Properties Through Stat3 Pathway. Oncotargets Ther 2020;13:1355-1363.
https://doi.org/10.2147/OTT.S229065 |
| 16 | Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li WC, Polotskaia A, Bissell MJ, Osborne TF, Tian B, Lowe SW, Silva JM, Borresen-Dale AL, Levine AJ, Bargonetti J, Prives C: Mutant p53 Disrupts Mammary Tissue Architecture via the Mevalonate Pathway. Cell 2012;148:244-258.
https://doi.org/10.1016/j.cell.2011.12.017 |
| 17 | Li R, Li SL, Shen L, Li JH, Zhang D, Yu JM, Huang LX, Liu N, Lu HW, Xu M: LINC00618 facilitates growth and metastasis of hepatocellular carcinoma via elevating cholesterol synthesis by promoting NSUN2-mediated SREBP2 m5C modification. Ecotox Environ Safe 2024;285
https://doi.org/10.1016/j.ecoenv.2024.117064 |
| 18 | Qiu T, Cao JW, Chen WZ, Wang JY, Wang YQ, Zhao LJ, Liu M, He LC, Wu G, Li HZ, Gu HH: 24-Dehydrocholesterol reductase promotes the growth of breast cancer stem-like cells through the Hedgehog pathway. Cancer Sci 2020;111:3653-3664.
https://doi.org/10.1111/cas.14587 |
| 19 | Angulo P, Kaushik G, Subramaniam D, Dandawate P, Neville K, Chastain K, Anant S: Natural compounds targeting major cell signaling pathways: a novel paradigm for osteosarcoma therapy. J Hematol Oncol 2017;10
https://doi.org/10.1186/s13045-016-0373-z |
| 20 | Hirotsu M, Setoguchi T, Sasaki H, Matsunoshita Y, Gao H, Nagao H, Kunigou O, Komiya S: Smoothened as a new therapeutic target for human osteosarcoma. Mol Cancer 2010;9
https://doi.org/10.1186/1476-4598-9-5 |
| 21 | Shahi MH, Holt R, Rebhun RB: Blocking Signaling at the Level of GLI Regulates Downstream Gene Expression and Inhibits Proliferation of Canine Osteosarcoma Cells. Plos One 2014;9.
https://doi.org/10.1371/journal.pone.0096593 |