Original Article - DOI:10.33594/000000816
Accepted 8 September 2025 - Published online
30 September 2025
SIRPα is An Inhibitory Receptor That Regulates NK Cell Activation and Function
bDepartment of Clinical Medicine, Aarhus University, 8220 Aarhus, Denmark,
cDepartment of Infectious Diseases, Aarhus University Hospital, 8220 Aarhus, Denmark,
dDepartment of Basic Dental Sciences, Faculty of Dentistry, Al-Ahliyya Amman University, 19328 Amman, Jordan,
eMedical Faculty Otto-von-Guericke, University of Magdeburg, 39120 Magdeburg, Germany,
fLaboratory of Experimental Medicine and Pediatrics, Faculty of Medicine and Health sciences, University of Antwerp, Belgium
Keywords
Abstract
Background/Aims:
Signal regulatory protein alpha (SIRPα) is an inhibitory receptor expressed on macrophages and dendritic cells. Recent cancer research studies have reported evidence of upregulation of SIRPα on natural killer (NK) cells. The present study aimed to investigate the role of SIRPα in NK cells during viral infection.Methods:
We utilized SIRPα knockout mice (SIRPα–/–) and lymphocytic choriomeningitis virus (LCMV) infection to examine the role of SIRPα in NK cells. Flow cytometry, in vivo killing assays, and molecular analyses were performed to assess NK cell activation, cytotoxic function, and associated signaling pathways.Results:
SIRPα expression was induced on NK cells during LCMV infection. The absence of SIRPα in knockout mice resulted in an increased proportion and activation of NK cells, with enhanced expression of cytotoxic markers and augmented NK cell-mediated killing of target cells. Mechanistically, loss of SIRPα was associated with downregulation of Src homology region 2–containing protein tyrosine phosphatase-1 (SHP-1) in NK cells. Importantly, SIRPα deficiency led to concomitant loss of CD8+ T cells and impaired viral control. In vivo killing assays indicated that activated NK cells mediated CD8+ T cell depletion in SIRPα–/– mice. Experimental NK cell depletion in these mice partially restored T cell immunity, reduced immunopathology, and improved viral clearance.Conclusion:
Our findings identify SIRPα as a critical inhibitory receptor that regulates NK cell effector functions. Loss of SIRPα unleashes NK cell activity but results in CD8+ T cell depletion and impaired antiviral immunity, highlighting the dual role of SIRPα in balancing NK cell activation and adaptive immune responses.Introduction
Natural killer (NK) cells are the main effector lymphocytes of the innate immune response that plays a crucial role as a first line defense during infection and cancer [1, 2]. An effective NK cell activation and cytotoxicity are orchestrated by a well-balanced surface expression of both activating and inhibitory receptors [3, 4]. During viral infection, NK cells can be activated by IFN-α and thus triggering their cytotoxic function [5]. However, NK cell activation can also result from the interaction of antigens with activating ligands or receptors such as NKG2D, NKp30, NKp44 and NKp46 [6-8]. One of the mechanisms of inhibitory receptor-mediated suppression of NK cell is via the recruitment and activation of Src homology 2 domain containing phosphatases, SHP-1 and SHP-2 [9, 10]. Activated SHP-1 and SHP-2 dephosphorylate immunoreceptor tyrosine-based inhibitory motifs (ITIMs), thereby preventing downstream activation signaling. SHPs set the threshold of NK cell activity and lack of SHPs leads to hyperresponsive NK cells [10-13].
Signal regulatory protein alpha (SIRPα) is one of the receptors whose interactions with its ligand (CD47) leads to the recruitment and phosphorylation of SHP-1 and SHP-2 [14, 15]. In phagocytic cells, SIRPα has been shown to interact with CD47, an integrin glycoprotein that is expressed in all cells. The CD47-SIRPα interaction leads to the upregulation, activation and recruitment of the protein tyrosine phosphatases, SHP-1 and SHP-2 [16]. In macrophages, SHP-1-mediated dephosphorylation inhibits myosin IIA activation necessary for phagocytosis [17, 18]. In addition to its inhibitory role in phagocytosis, ligation of SIRPα has also been shown to inhibit dendritic cells (DC) survival and activation. Thus, DCs lacking SIRPα expression display enhanced production of IL-12, higher expression of co-stimulatory molecules, and enhanced antigen presentation function resulting in increased T cell response [19-21]. The role of SIRPα on macrophages and DCs contrasts its role on CD8+ T cells [22]. SIRPα expression on subset of CD8+ T cells is associated with proliferation, IFN-γ production and cytotoxic activity [22].
CD47-SIRPα expression and interaction on NK cells have recently attracted lot of attention. NK cells have been reported to express the highest level of CD47 at both mRNA and protein level compared to other immune cells [23, 24]. However, the lack of CD47 resulted in an increased expression of NK cells effector function and interferon stimulated genes [24, 25] CD47, a widely known antiphagocytic molecule, is now reported to be a crucial regulator of NK cell function [25]. In addition, CD47 ligand thrombospondin-1 (TSP1) is reported to be expressed on NK cells and inhibit NK cells proliferation, activation and function [26]. Mechanistically, CD47-TSP1 suppresses NK cell IFN-γ production by activating the JAK/STAT3 pathway [27]. Recent studies have now added SIRPα to the list of inhibitory receptors on NK cells [28, 29]. SIRPα is upregulated on NK cells upon IL-2 stimulation [28] and the presence of SIRPα on NK cells prevent NK cell mediated killing of cancer cells [28, 29]. Therefore, SIRPα and its counterparts (CD47 and TSP1) have cell-intrinsic roles in regulating NK cells activation and function.
In this current study, we show evidence that SIRPα is expressed on NK cells during viral infection using LCMV model. Experiments on genetically deficient (SIRPα-/-) mice demonstrate that absence of SIRPα led to increased NK cell proportion, activation and cytotoxicity activities on target cells. Consequently, lack of SIRPα in SIRPα-/- mice led to abortive CD8+ T cell response due to NK cell mediated killing and subsequent diminished virus control. Depletion of NK cells in the SIRPα-/- mice partially restored CD8+ T cell responses, ameliorated immunopathology and enhanced virus clearance. Our results provide important insight that SIRPα is an essential immune checkpoint molecule with regulatory function on NK cells.
Materials and Methods
Mice
Inbred B6.129P2-Sirpatm1Nog/Rbrc (SIRPα-/-) and C57BL/6J (WT) mice were purchased
from
Jackson Laboratory. P14 mice expressing the LCMV-GP33-41-specific TCR as a transgene were originally
obtained
from Prof. Tak W. Mak (The Campbell Family Cancer Research Institute and University Health Network,
Toronto,
Canada). P14 mice are crossed with CD45.1 congenic mice to track lymphocytes during adoptive transfer
experiments. All experiments were performed using mice older than 8 weeks of age, housed in ventilated
cages and
the health status of the mice was checked daily. All animal experiments were approved under license number
(84-02-04.2013.A242) by Landesamt fur Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen. All animal
care
and use protocols adhere to national (Tierschutzgesetz) and European (Directive 2010/63/EU) laws and
regulations
as well as European Federation of Animal Science Associations (FELASA). Animals were euthanized using
cervical
dislocation methods.
Virus
The LCMV-WE and LCMV-docile virus stocks used in this study were originally obtained from Prof. F.
Lehmann-Grube, Heinrich Pette Institute, Hamburg, Germany and was expanded on L929 cells (obtained from
ATCC,
NCTC clone 929). LCMV-Docile was provided by Prof. Dr. R. Zinkernagel (University of Zurich, Zurich,
Switzerland) and was propagated on L929 cells.
Flow cytometry analysis of NK cells
First, splenocyte from LCMV infected C57BL/6J mice were used to analyze SIRPa surface expression on immune
cells. Splenocytes were simply washed and stained with mouse anti-CD172a (P84, 144011), CD3e (17A2,
47-0032-82),
CD8 (53-6.7, 47-0081-82), CD19 (eBio1D3, 47-0193-82), CD11c (N418), CD11b (M/70) and CD4 (GK1.5) at 4 °C
for 30
min and then washed with FACS buffer and analyzed using flow cytometry. For analysis of NK cell surface
expression markers, splenocytes from WT and SIRPa knockout mice were harvested, lysed with 1 mL BD lysing
solution (BD Biosciences) and washed with FACS buffer. Mouse-specific antibody to NK1.1(PK136,
25-5941-82), CD44
(IM7, 563736), NKG2D (62-5882-82), CD69 (H1.2F3, 561238), KLRG1(2F1, 740553), TCR-β (H57-597, 47-5961-82),
NKp46
(29A1.4, 11-3351-82) were used. Splenic cells were stained with antibodies and incubated at 4 °C for 30
min and
then washed with FACS buffer via centrifugation at 1500rpm for 5 min. Cells were re-suspended in FACS
buffer and
were analyzed. To analyze the intracellular cytokines, splenocytes were homogenized, and cells were then
stained
with surface marker NK1.1(PK136, 25-5941-82), TCR-β (H57-597, 47-5961-82), TRAIL (17-5951-82), then fixed
with
2% formaldehyde solution in PBS for 10 min, permeabilized with 1% saponin solution, and then stained with
anti-granzyme B (NGZB, 12-8898-82), perforin (eBioOMAK-D,11-9392-82), IFN-γ (XMG1.2, 17-7311-82),
antibodies.
For Lamp-1 staining, anti-CD107a antibody was added for the 5-hr incubation period, and the
immunofluorescence
was measured after additional staining with anti-NK1.1 antibody. FACS data were analyzed with the FlowJo
software (FlowJo LLC, Ashland, OR, USA).
NK cells culture and cytotoxicity assay
Using a mouse NK cell isolation kit (130-115-818; Milteny Biotec, Germany), NK cells were negatively
sorted from
naïve or LCMV infected (WT and SIRPa-/- mice) 24 hours after infection with 2x106 PFU LCMV-WE.
The NK
cell negative sorting was performed according to the manufacturer’s protocol. For cytotoxicity assay,
labeled
RMA/S and RMA cells with 10 μM Cell Proliferation Dye eFluor 450 (Invitrogen, 65-0842-85) were cocultured
with
isolated NK cells at different effectors/targets ratios. NK cells were co-cultured with RMA or RMA-S cells
in
complete RPMI 1640 and supplemented with 10mM HEPES (pH 7.2), 2mM L-glutamine, 105 U/L
Penicillin, 0.1 g/L Streptomycin, and 10% FCS. The co-cultures were maintained in incubators at 37°C, 5%
CO2. After 18 hours of incubation, 7-AAD (Invitrogen, 00-6993-50) was added, and the percentage
of
7-AAD+ cells among target cells was measured by flow cytometry.
Plaque assays
LCMV viral titers were detected by plaque-forming assays on MC57 fibroblasts (obtained from by the Ontario
Cancer Institute, Canada) cultured at 37°C in Dulbecco’s modified Eagle medium (DMEM) containing 2% fetal
calf
serum (FCS) and 1% penicillin/streptomycin. Organs were smashed and plasma was diluted in DMEM containing
2%
FCS, titrated 1:3 over 12 steps, and incubated on MC57 cells. After 4 hours of incubation at 37°C, an
overlay
(1:1 mixture of methyl cellulose and Iscove’s Modified Dulbecco’s Medium) was added and the virus
preparation
was further incubated for 48 hours followed by staining of LCMV plaques. For staining, cells were fixed
with 4%
formaldehyde solution in phosphate-buffered saline (PBS), permeabilized with a 1% Triton-X solution in
PBS,
blocked with 10% FCS in PBS, and stained with anti-LCMV nucleoprotein (NP) antibody (made in house).
Enhanced
chemiluminescence (ECL)-conjugated anti-rabbit IgG antibody was used as a secondary antibody. Plaques were
detected by color reaction (0.2 M Na2HPO4 + 0.1 M citric acid + 30%
H2O2 + o-phenylenediamine dihydrochloride). All chemicals were purchased from
Sigma-Aldrich (Germany).
Mixed Bone marrow chimera
Female C57BL/6J mice at least 8-10 weeks old were irradiated with a total of 1050 rad. After 24 hours,
bone
marrow from donor mice (WT, SIRPa KO and CD45.1) was isolated under sterile conditions. The bone marrow
chimera
mice were intravenously reconstituted with 1:1 composition of bone marrow from WT
CD45.1+:CD45.2+ SIRPα+/+ mice in one group, and WT
CD45.1+:CD45.2+ SIRPα-/- mice in another group. Mice
were
analyzed 35 days after reconstitution. Recipient mice were infected with LCMV for 24 hours, NK cells
response
was analyzed using flow cytometry.
Immunoblotting
NK cells were dissociated and lysed in sodium dodecyl sulfate (SDS) buffer (1.1% SDS, 11% glycerol,
0.1M
Tris; pH 6.8) plus with 10% 2-mercaptoethanol (Sigma). Total cell extracts were loaded in 10%
SDS-polyacrylamide
electrophoresis gels and transferred onto the PVDF membrane (GE). After electrophoresis, membranes were
blocked
in 5% bovine serum albumin (-) in PBS supplemented with 1% Tween-20 for 1 h at RT and then incubated
overnight
at 4 ℃ with the following antibodies: anti-SHP (Syk) (Y352/Y319), anti-GAPDH (CST). The membrane was
incubated
with conjugated HRP-anti-rabbit IgG antibody at RT for 1h. The image was developed with the Bio-Rad
ChemiDoc
imaging system (Bio-Rad Laboratories) and analyzed with the Bio-Rad software.
Tetramer, surface, intracellular staining, and flow cytometry analysis
The LCMV-specific CD8+ T cell response upon LCMV infection was detected with a tetramer complex
of
major histocompatibility complex (MHC) class I (H-2Db) and LCMV GP33-41 (KAVYNFATM)
peptide Tetramers were provided by the Tetramer Facility of the National Institutes of Health (NIH;
Bethesda,
MD, USA). Cells were stained with allophycocyanin-labeled GP33 (GP33/H-2Db) tetramers for 15 min at 37°C.
After
incubation, the samples were stained with monoclonal antibody to CD8a for 30 minutes at 4°C. Absolute
numbers of
GP33-specific CD8+ T cells were counted by fluorescence-activated cell sorting (FACS) with
calibrating beads (340486; BD Bioscience, Germany).
For surface staining, erythrocytes were lysed with 1 ml BD lysing solution (BD Biosciences) or smashed
splenocytes from LCMV-infected mice washed once with fluorescence-activated cell sorting (FACS) buffer and
analyzed by flow cytometry. Absolute numbers of cells were calculated based on results from fluorescent
calibrating beads. Surface antibodies: CD8a (53-6.7), CD4 (GK1.5), CD11c (N418), MHC class II
(M5/114.15.2), and
CD80 (16-10A1), were purchased from Thermo-fisher. Antibodies CD11b (M/70), CD44 (IM7) and CD86 (GL1)
purchased
from BD Biosciences. Stained cells were acquisitioned on BD LSRFortessa™ cell analyzer (BD Bioscience),
and data
were analyzed with the FlowJo software (FlowJo LLC, Ashland, OR, USA).
For intracellular cytokine staining, smashed splenocytes from LCMV-infected mice were cultured in 5% FCS
DMEM
medium supplemented with LCMV GP33-41 peptide (5 µg/ml) for 1 hour at 37°C in an incubator.
After 1
hour, brefeldin A (25 µg/ml; B7651; Sigma, Germany) was added, and the cells were incubated for another 4
hours
at 37°C. After a total of 5 hours, splenocytes were washed with FACS buffer, stained for surface
anti-mouse CD8
antibody at 4°C for 30 minutes and then fixed with 2% formalin in PBS at room temperature for 10 minutes.
After
another washing step, cells were incubated for intracellular staining with antibodies to IFN-γ (XMG 1.1,
eBioscience) in 0.1% saponin (S4521; Sigma) in FACS buffer for 30 minutes at 4°C, washed, and analyzed
with flow
cytometry.
Adoptive Transfer of P14 CD8+ T cells
CD8+ T cells were bead purified from splenocytes of CD45.1+P14 mice using
CD8+
T cell isolation kit (130-104-075, Miltenyi Biotec) and CD8+ T cells were obtained using
procedure
according to the manufacturer. Naïve CD8+ T cells from CD45.1 x P14 mice were transferred
intravenously to WT and SIRPa deficient mice. The recipient mice were then infected with LCMV and total
CD45.1
P14 CD8+ T cells were analyzed using flow cytometry.
NK cell depletion
NK cells were depleted with i.p injection of 200μg anti-NK1.1 Ab (PK136, BioXcell) or IgG2a isotype
control at
day -3 and day 1 post infection. Cell subset depletions were confirmed by FACS analysis to be greater than
95%.
In vivo killer assay
Naïve 106 CD8+ T cells from spleens of P14 x CD45.1 mice were negatively sorted by
CD8+ T cell isolation kit (Milteny Biotec). These cells were transferred to C57BL/6J mice at
day -1
and intravenously infected with 200 PFU of LCMV-WE. After 5 days, 1 x 106 negatively MACS
sorted
total CD8+ T cells from these mice were transferred to NK cell depleted
SIRPα+/+ or SIRPα-/- mice which were already
i.v
infected with 200 PFU of LCMV-WE 3 days before. After 4 hours of transfer and in vivo incubation in
recipient mice, the spleen of recipient mice was collected and the total number of P14 were analyzed by
FACS.
Liver enzyme activity measurements
For quantification of Alanine amino transaminase (ALT), aspartate transaminase (AST) activity, serum
samples
were sent to the central diagnostic laboratory of the University Hospital.
Statistical analysis
Data are depicted as means ± S.E.M. Unpaired Student’s t-tests were used to detect statistically
significant
differences between groups. P values lower than 0.05 were considered statistically significant.
Statistical
analyses and graphical presentations were computed with Graph Pad Prism, version 10.03 (GraphPad Software,
USA).
Results
SIRPα expression inhibits NK cell activation
Following previous studies highlighting the expression and upregulation of SIRPα on NK cells in cancer
[28, 29],
we opted to determine the expression of SIRPα on NK cells during viral infection. C57BL/6 mice were
infected
with 2x106 PFU LCMV-WE intravenously (i.v) for 24 hours, SIRPα surface expression on innate and
adaptive immune cells was analyzed. We found that SIRPα is expressed on activated NK cells and other
immune
cells after 24 hours (Fig. 1A). Next, we evaluated whether SIRPα is upregulated after 7 days post LCMV
infection. Unsurprisingly, we observed an upregulation of SIRPα across all immune cells (Supplementary
Fig. 1A).
Notably, an ongoing immune response to viral infection will lead to continuous production of IL-2 that was
reported to upregulated SIRPα on NK cells [28, 29]. To further investigate whether SIRPα plays a role in
NK cell
response, wild type (WT) and SIRPα-/- mice were either left naive or infected
with
2x106 PFU LCMV-WE for 24 hours, splenocytes were analyzed by flow cytometry. Compared to WT
mice, we
observed significantly increased proportion of NK cells in spleen of both naive and LCMV infected
SIRPα-/- mice (Fig. 1B). Next, we evaluated the expression levels of NK cell
activating
markers and found that NK cells from SIRPα-/- mice displayed a significantly
higher
NKG2D expression (Fig. 1C), CD69 expression (Fig. 1D), KLRG1 expression (Fig. 1E) and NKp46 expression
(Fig. 1F)
compared to WT mice. Together, our results provided evidence of SIRPα expression on activated NK cells
following
a viral infection and deficient of SIRPα led to enhanced NK cells activation.
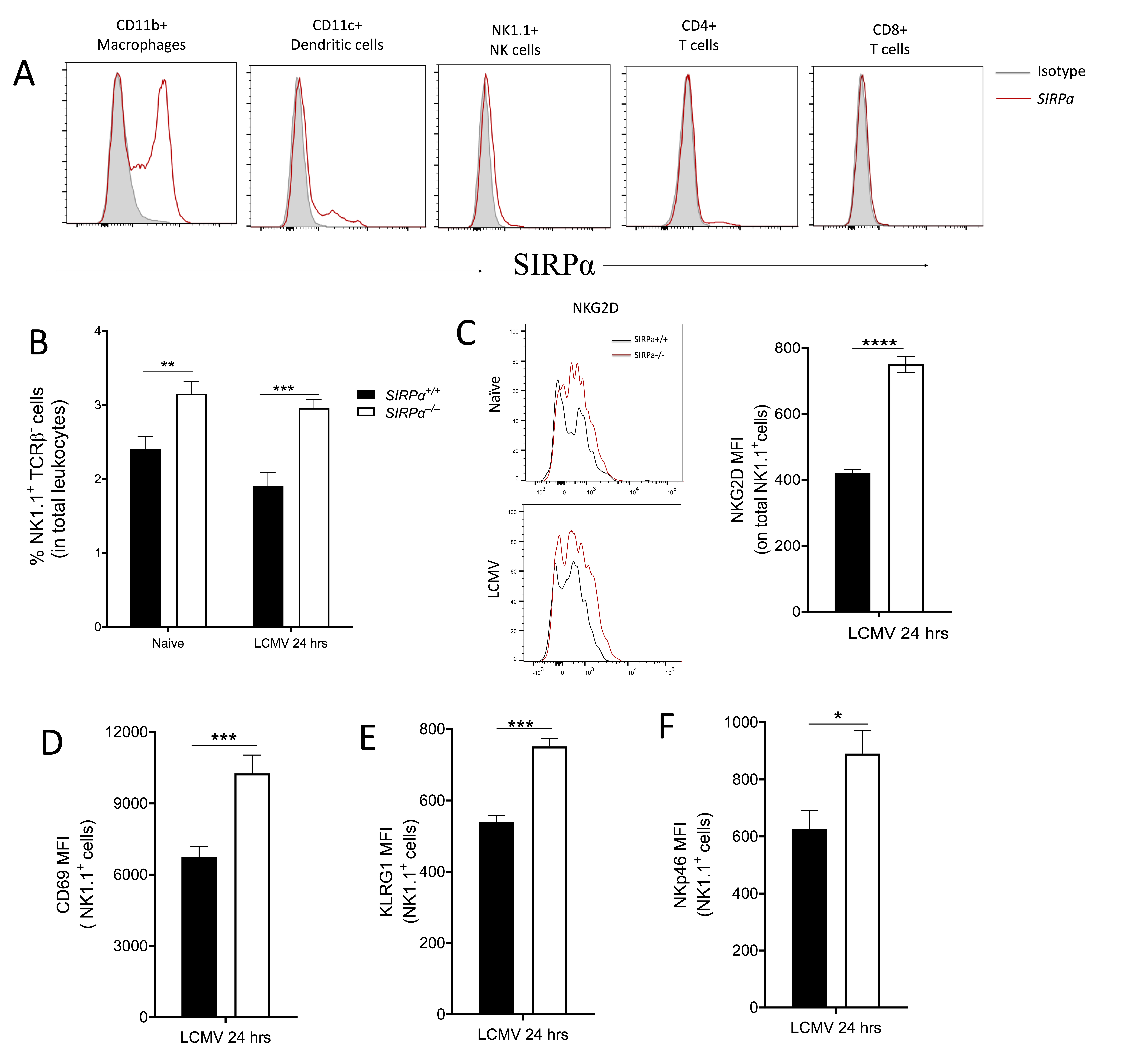
Fig. 1: SIRPα expression inhibits NK cell activation. WT and SIRPα-/- mice were intravenously infected with 2x106 PFU LCMV-WE. Mice were put to death after 24 hours of infection. (A) SIRPα cell surface expression on different splenic immune cell analyzed by flow cytometry in infected WT mice. (B) Shown is the percentage of NK1.1+ TCRβ- cells in the splenocytes harvested from naïve and infected WT and SIRPα-/- mice. (C) Representative histogram for the NKG2D expression on the splenic NK cells retrieved from infected WT and SIRPα-/- mice (left panel). In the right panel, the show is median fluorescence intensity (MFI) for the same experiment. (D) The bar graph depicts the expression of NK cell activation marker MFI CD69, (E) MFI KLRG1 (F) and MFI NKp46. The data shown was confirmed in two independent experiments (n=5) and are shown as mean ± SEM. Significant differences between the two groups were detected by Student’s t-test and are indicated as follows; (ns, not significant; * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001).
Absence of SIRPα enhances NK cell mediated cytotoxicity
To evaluate the impact of SIRPα on NK cell cytotoxic function, we infected WT and SIRPα knockout mice with
LCMV
and assessed NK cell cytotoxicity markers. SIRPα-/- mice exhibit a
significantly
higher percentage of Perforin+ NK cells (Fig. 2A), IFNγ+ NK cells (Figure2B) and
Granzyme
B+ NK cells (Fig. 2C). We further observed that lack of SIRPα resulted in significantly
enhanced NK
cell degranulation as measured by increased CD107a expression (Fig. 2D). Additionally, we observed a
significantly higher expression of tumor necrosis factor–related apoptosis-inducing ligand (TRAIL)
(Fig.
2E) in SIRPα-/- mice. With the observed increased expression of these
cytotoxic
signatures, we opted to evaluate the NK cell mediated killing of target cells in WT and SIRPα deficient
mice.
Here, we used RMA cell line that has a sufficient surface expression of major histocompatibility complex
class I
molecules (MHC-I) and RMA-S cell line that express deficient MHC-I. Both RMA and RMA-S cells were labeled
with
dye and co-cultured with NK cells isolated from naïve and LCMV infected WT and
SIRPα-/- mice. We found that the co-culture of RMA or RMA-S with naïve WT
and
SIRPα-/- NK cells does not lead to any cytotoxic difference (Supplementary
Fig.
1B). However, when we exposed RMA and RMA-S with activated NK cells isolated from LCMV infected WT and
SIRPα
knockout mice, we found a significant increased NK cell mediated lysis of target cells among
SIRPα-/- NK cells (Fig. 2F). Next, we investigated whether the observed NK
cell
phenotype and function can suppress early LCMV replication. We infected WT and SIRPα-/-
mice with LCMV for 24 hours and evaluated the viral load. We found no difference in LCMV viral
load (Supplementary Fig. 1C). These findings suggest that the absence of SIRPα on NK cells resulted into
an
increased expression of NK cell cytotoxic markers and an enhanced NK cell mediated lysis of target cells.
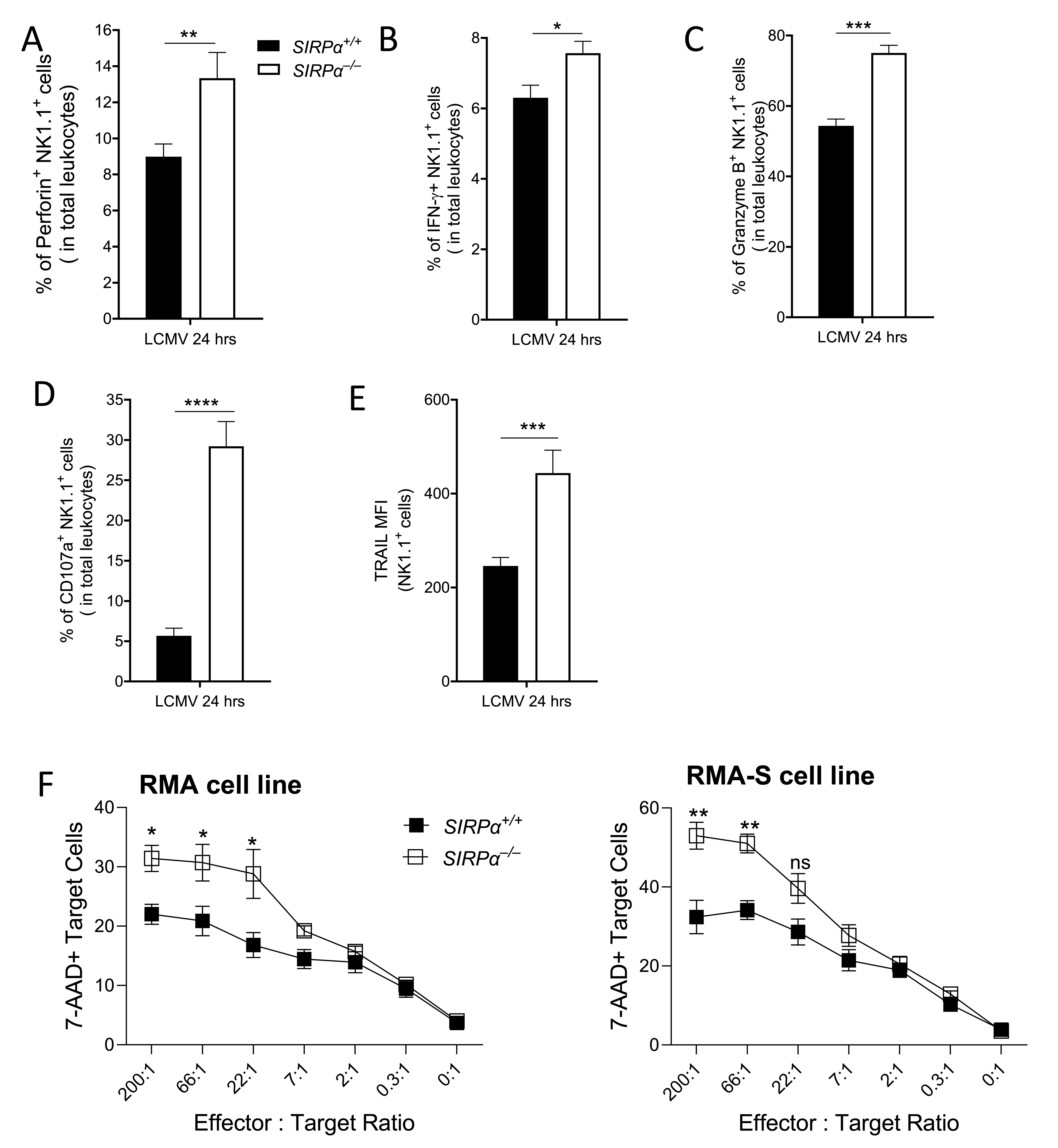
Fig. 2: Absence of SIRPα enhances NK cell mediated cytotoxicity. WT and SIRPα-/- mice were intravenously infected with 2x106 PFU LCMV-WE. Mice were put to death after 24 hours of infection, and the effector functions of NK cells were analyzed as indicated. (A) Frequency of perforin-producing NK cells of splenocytes retrieved from infected WT and SIRPα-/- mice. (B) Percentage of IFN-γ - producing NK cells of splenocytes retrieved from infected WT and SIRPα-/- mice. (C) Percentage of Granzyme B- producing NK cells of splenocytes retrieved from infected WT and SIRPα-/- mice. (D) Expression of degranulation marker (CD107a) on splenic NK cells from infected WT and SIRPα-/- mice. (E) Surface expression of TRAIL on splenic NK cells from infected WT and SIRPα-/- mice. (F) Shown is the NK cell–mediated killing to target cells (RMA and RMA-S) as measured by expression of 7-AAD+ on target cells after co-cultured at different ratios with NK cells harvested from infected mice. The data shown was confirmed in two independent experiments (n=5) and are shown as mean ± SEM. Significant differences between the two groups were detected by Student’s t-test and are indicated as follows; (ns, not significant; * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001).
Deficiency of SIRPα is intrinsically linked to downregulation of SHP-1 on NK cells
To determine whether SIRPα exerted an intrinsic or extrinsic effect on NK cells during LCMV infection, we
created a mixed bone marrow chimeras using CD45.1-marked, SIRPα WT and SIRPα-/-
mice
(CD45.2). C57BL/6 mice were irradiated and after 24 hours, we adoptively transferred 107 bone
marrow
cells from CD45.1 with SIRPα WT or CD45.1 with SIRPα-/- mice in a 50:50 ratio.
After 35
days of reconstitution, recipient mice were infected with LCMV and NK cells were analyzed 24 hours as
illustrated in (Supplementary Fig. 1C). Donor SIRPα-/- NK cells from the chimeras
exhibited increased expression of NKG2D activating marker compared to WT donor NK cells (Fig. 3A). Further
analysis of other NK cells effector function reveals that lack of SIRPα on NK cells results in a
significantly
higher percentage of CD107a+ NK cells (Fig. 3B) and IFN-γ+ NK cells (Fig. 2C). These
findings suggest that SIRPα mediates an NK cells intrinsic effect during LCMV infection. However, the
SIRPα-CD47
interaction is well known to recruit and activate Src homology 2 domain- containing phosphatases SHP-1 and
SHP-2. These SHPs are known to induce an inhibitory effect on NK cells activation (30, 31). We isolated NK
cells
from WT and SIRPα deficient mice and analyzed SHP-1 level using western blot. The lines 1 and 2 and from
two WT
mice, while line 3 and 4 are from two Knockout mice. We found that the deficiency of SIRPα on NK cells led
to
downregulation of SHP-1(Fig. 2D). Our results reveal that SIRPα mediate an intrinsic effect on NK cells
and the
lack of SIRPα leads to downregulation of SHP-1.
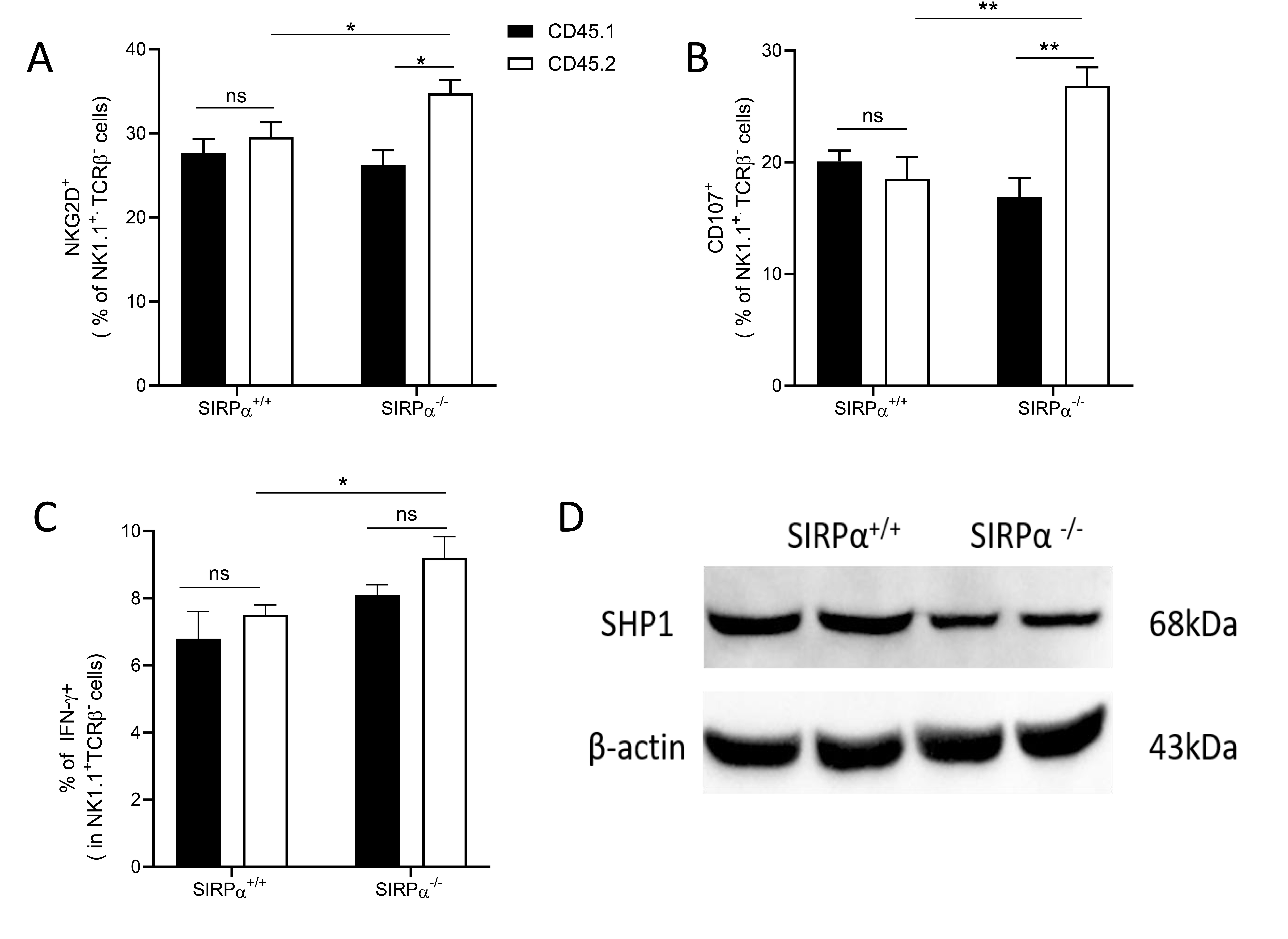
Fig. 3: Deficiency of SIRPα is intrinsically linked to downregulation of SHP-1 on NK cells. C57BL/6 mice were radiated and reconstituted with 107 (1:1 composition of) bone marrow cells from CD45.1+:SIRPα+/+ or CD45.1+:SIRPα-/- mice. On day 35 after bone marrow reconstitution, these mice were intravenously infected with 2x106 PFU of LCMV-WE for 24 hours. NK cell responses were analyzed from the spleen of recipient mice. (A) Percentage of NKG2D expression on NK cells, (B) percentage of CD107a+, and (C) percentage of IFN-γ+ NK cells. (D) SHP-1 protein expression of NK cells from LCMV infected WT and SIRPα -/- mice were analyzed by immunoblotting. The data shown was confirmed in two independent experiments (n=5) and are shown as mean ± SEM. Significant differences between the two groups were detected by Student’s t-test and are indicated as follows; (ns, not significant; * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001).
Lack of SIRPα limits antiviral CD8 T cell immunity and aggravates virus control.
Since antiviral CD8+ T cell responses play an indispensable role in the clearance and control
of
acute and persistent LCMV infections [5, 32], we examined the impact of the observed NK cell phenotype on
antiviral CD8+ T cells response. We infected WT and SIRPα-/- mice with
LCMV-WE
strain and analyzed T cell responses as well as virus control. The LCMV-specific CD8+ T cell
response
was evaluated using a tetramer complex of major histocompatibility complex (MHC) class I
(H-2Db) and
LCMV GP33-41 (KAVYNFATM) peptide Tetramers [33]. We found that the lack of SIRPα resulted in a
significantly reduced number of total CD8+ T cells (Fig. 4A) and virus specific CD8+
T
cells (Fig. 4B) in blood at indicated time points. Splenic analysis of CD8+ T cell activity as
demonstrated by the percentage of IFN- producing CD8+ T cells showed significantly reduced
CD8+ T cell effector functions in SIRPα-/- mice compared to WT mice (Fig.
4C). The
LCMV-WE strain used in this experiment induces an acute infection and its clearance crucially depends on
CD8+ T cells [34]. Depending on the dose of the viral inoculum, LCMV-WE clearance is observed 8
to 12
days post infection [5]. However, due to the impaired CD8+ T cells response in the
SIRPα-/- mice, these mice failed to control the virus even after 12 days post infection.
The
virus titers were significantly higher in serum and several organs of the SIRPα-/- mice
while
effective virus clearance was observed in WT mice (Fig. 4D). Next, we employed the LCMV-docile which
induces a
chronic infection (34). The infection of WT and SIRPα-/- mice with LCMV-docile exhibited
similar patterns as observed in the acute infection. Reduced CD8+ T cells (Supplementary Fig.
2A-B)
and higher virus titers in all organs (Supplementary Fig. 2C) were apparent in the
SIRPα-/-
compared to WT mice. Next, we investigated a possible potential mechanisms of the reduced T cells response
in
the SIRPα knockout mice. As SIRPα is mainly expressed on antigen presenting cells, and these cells
are
crucial for the priming and activation of CD8 T cells, we analyzed the activation of dendritic cells
during LCMV
infection. We found an increased activation as mirrored by CD80 and CD86 expression levels on CD11c DCs in
the
SIRPα-/- compared to WT mice (Fig. 4E). These results seem quite controversial because
an
increased antigen presentation markers on DC are associated with increased T cell response [35]. This
observation suggests that there is another potential mechanism that may partly explain the reason for
reduced T
cell response in SIRPα deficient mice. We investigated other possible underlying mechanisms. Since it is
well
established that NK cells can regulate CD8+ T cells in the context of viral infections by
cytotoxic
killing [36, 37], we investigated the NK cell-mediated killing of CD8+ T cells in WT and
SIRPα-/- mice. To this end, we transfer CD45.1-positive P14 CD8+ T cells into
WT
and SIRPα-/- mice and subsequently infected with LCMV-WE. Flow cytometry analyses
revealed a
significantly reduced expansion of the transferred P14 CD8+ T cell in the
SIRPα-/-
mice (Fig. 4F) suggesting an enhanced killing of transferred P14 CD8 T cells. Overall, our data indicate
that
lack of SIRPα in the SIRPα-/- mice leads to significantly reduced endogenous and
exogenous CD8 T cells and aggravates LCMV infection.
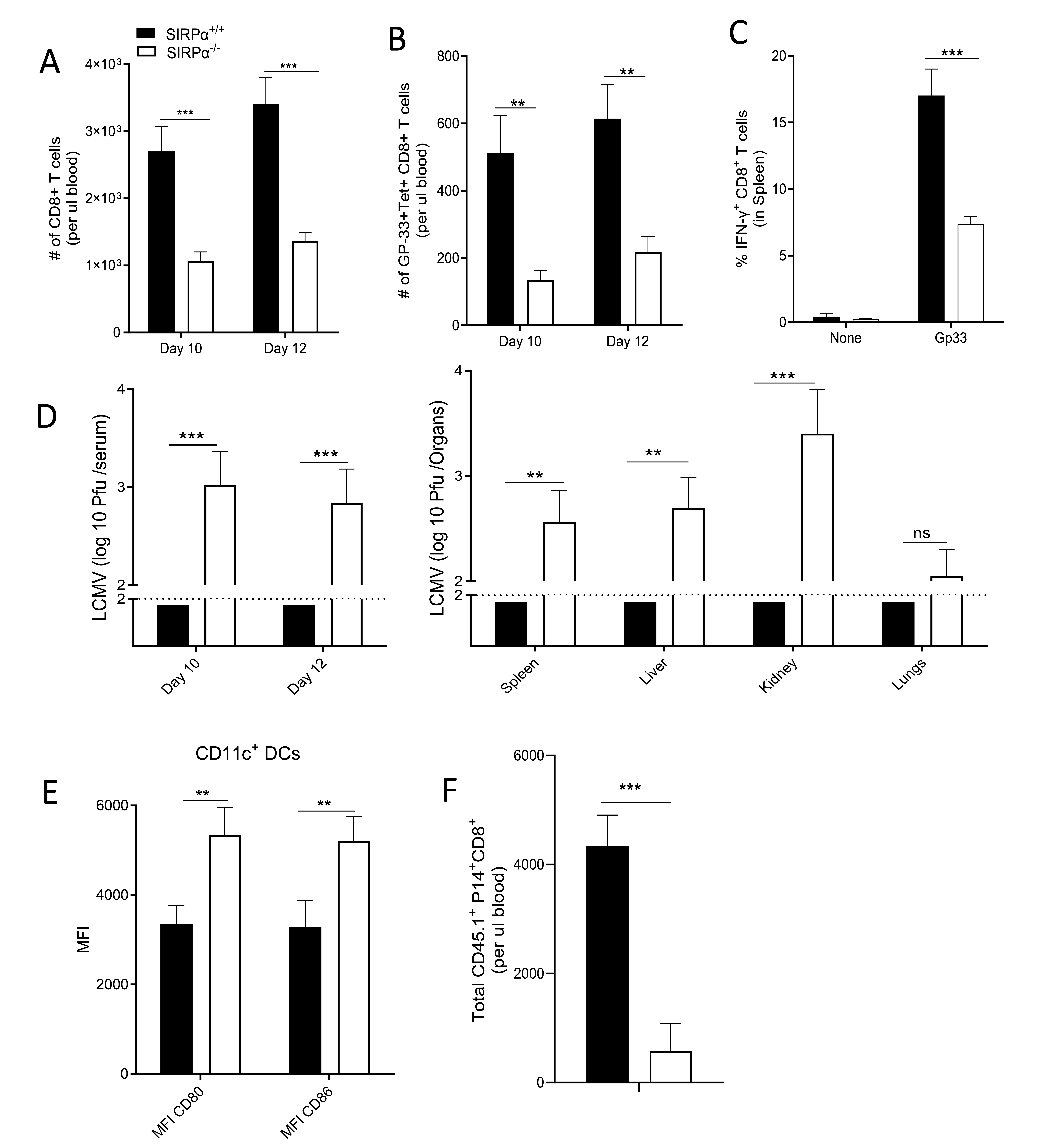
Fig. 4: Lack of SIRPα limits antiviral CD8+ T cell immunity and aggravate virus control. WT and SIRPα-/- mice were intravenously infected with 2x106 PFU LCMV-WE and flow cytometry analysis was performed in the blood and splenocytes at indicated time points. (A) absolute number of CD8+ T cells and (B) absolute number of GP33-Tet+ CD8+ T cells in blood at day 10 and 12 post infection. (C) Intracellular cytokine staining of IFN-γ + CD8+ T cells in spleen after 12 days of infection. (D) Bar graph depicts the LCMV titter in serum and viral load in spleen, liver, kidney and lung at 12 days post infection. (E) Splenic APC activation from infected WT and SIRPα-/- mice as measured by mean fluorescence intensity (MFI) of CD80 and CD86 on CD11c+ DC cells. (F) Isolation and adoptive transfer of 104 naïve CD45.1+ P14+ CD8+ T cells to SIRPα+/+ or SIRPα-/- mice on day -1. The recipient mice were infected with 2x104 PFU LCMV WE. Absolute number of CD45.1+CD8+ T cells at 8 days post infection. Data shown are means ± SEM. Significant differences between the two groups were detected by Student’s t-test and are indicated as follows; (ns, not significant; * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001).
Lack of SIRPα promotes NK cell mediated killing of CD8 T cells
Several studies have shown that NK cells are crucial for the regulation of T cell response through NK cell
mediated killing of CD8+ T cells during viral infection [5, 32, 34, 36-40]. Based on these
reported
studies and our data derived in Fig. 4F, we hypothesized that SIRPα might be one of the regulators
involved in
shaping the NK cell activity against antiviral CD8+ T cells. To test this, we performed in
vivo killer assay to evaluate the NK cell-mediated killing of activated CD8+ T cells.
Naive
CD45.1+ P14 T cells were transferred into C57BL/6J mice and infected with LCMV for 5 days. At
-3-day,
WT and SIRPα knockout mice were treated with isotype or anti-NK cell depleting antibodies and then
infected with
LCMV or kept naive. At day 5, P14 CD8+ T cells were isolated and transferred into the infected
WT and
SIRPα-/- mice as shown in (Supplementary Fig. 3A-B). Splenic cells analysis after 4
hours of
adoptive transfer revealed a markedly increased percentage of NK cells in SIRPα-/-
compared to
WT mice and an effective NK depletion in anti-NK treated mice (Fig. 5A). We found that the transfer of
CD45.1+ P14 T cells to naïve WT and SIRPα-/- mice led to no significant
difference
in NK cell mediated killing (Supplementary Fig. 3C). Interestingly, an adoptive transfer of CD45.1+
P14 T cells to LCMV infected WT and SIRPα-/- mice led to diminished numbers of
transferred P14 T cells in the SIRPα-/- compared to WT mice (Fig. 5B). This result
suggests
that the increased number of activated NK cells in the SIRPα-deficient mice led to an enhanced cytotoxic
killing
of CD8+ T cells, thereby reducing the number of antiviral T cells. However, this data also
suggests
that there are other unknown factors since the depletion of NK cells does not fully stop CD8 T cells
reduction.
To further corroborate these findings, we depleted NK cells in WT and SIRPα-/- mice and
infected them with LCMV. After 8 days of infection, a FACS analysis of splenic cells showed a partial
restoration of the total number of CD8+ T cells (Fig. 5C) as well as the absolute number of
virus-specific CD8+ T cells (Fig. 5D) in the anti-NK-treated SIRPα-/- mice.
Additionally, we observed alleviated liver immunopathology as measured by ALT (Fig. 5E) and AST enzyme
levels
(Fig. 5F) in SIRPα-/- mice. Like the observed restoration of the antiviral CD8 T cells,
we
observed improved virus control in SIRPα-/- mice upon NK cell depletion (Fig. 5G). Our
results
suggest that the lack of SIRPα led to NK cell activation, increased NK cell mediated killing of
CD8+
T cells, and an exacerbated LCMV infection, which was partially rescued by the depletion of NK cells.
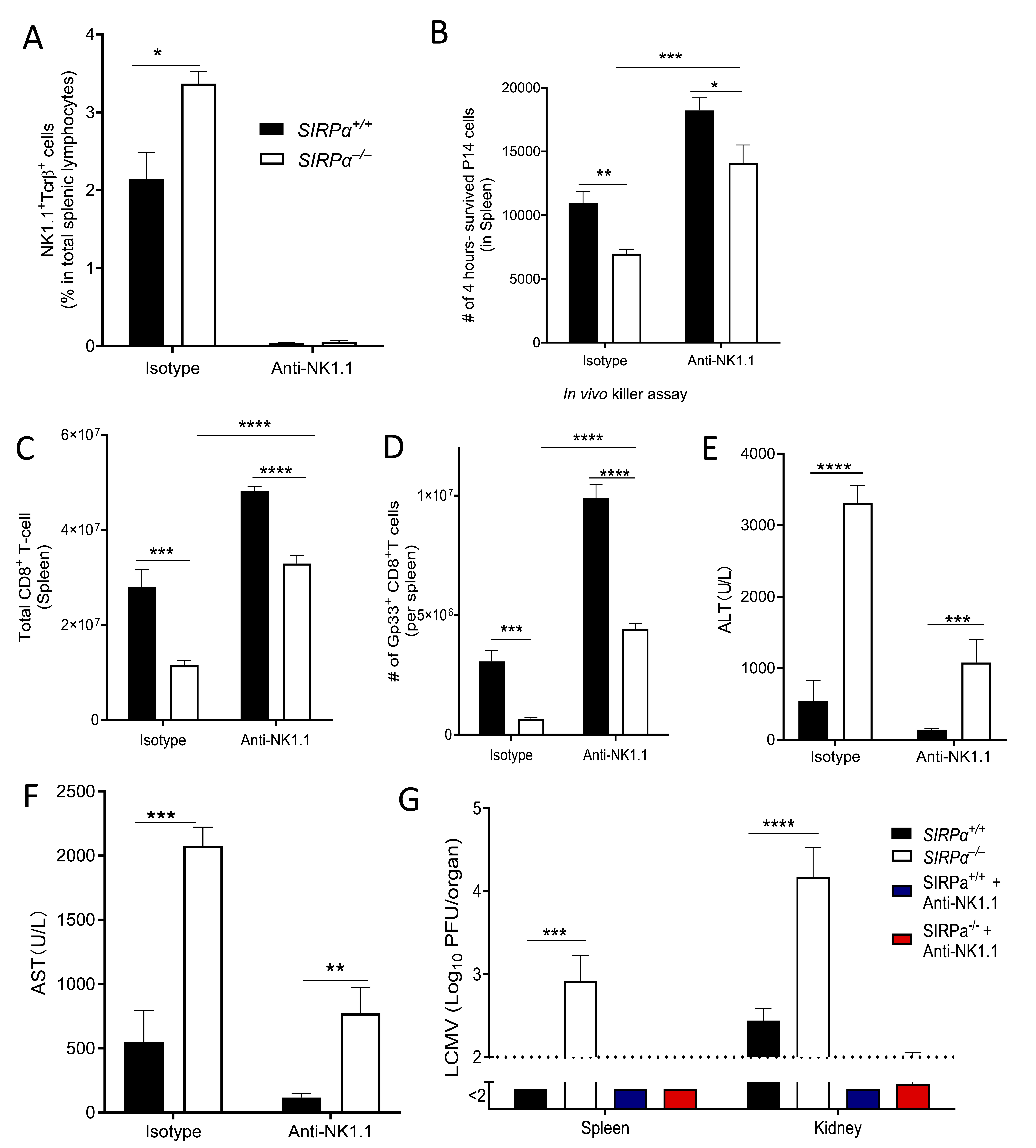
Fig. 5: Lack of SIRPα promotes NK cells mediated killing of CD8 T cells. In vivo killer assay experiment of the transfer of activated P14 cells into LCMV infected WT and SIRPα-/- mice for 3 days before transfer. (A) Percentage of NK cells in splenocytes of WT and SIRPα-/- mice after treatment with isotypes or Anti NK1.1 and (B) absolute number of transferred CD45.1+P14 cells. WT and SIRPα-/- mice were treated with 100µg of anti-NK1.1 or isotype control at day -3, day -1 and infected with 2x106 PFU LCMV-WE and flow cytometry analysis was performed at 8 dpi. (C) Total number of CD8+ T cells in spleen and (D) absolute number of GP33-Tet+ CD8+ T cells. (E) Levels of ALT and (F) levels of AST in serum. (F) The virus titters in the spleen and kidney of WT and SIRPα-/- mice after treatment with isotypes or anti-NK depleting antibodies. Data shown are means ± SEM. Significant differences between the two groups were detected by Student’s t-test and are indicated as follows; (ns, not significant; * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001).
Discussion
Previous studies investigating the role of SIRPα in different immune cells highlighted that the function of SIRPα critically depends on the type of immune cell. SIRPα elicits inhibitory functions among innate immune cells studied so far, while mediates activating functions on CD8+ T cells [15, 19, 20, 22, 41, 42]. SIRPα expression inhibits phagocytic activities in macrophages and limits DC activation and DC-dependent priming of CD8+ T cells [18, 19]. In clear contrast to its inhibitory role on innate immune cells, reports have showed an immune-stimulatory role of SIRPα by enhancing the proliferation, activation, and effector functions of T cells [22]. Our study further investigates the function of SIRPα to NK cells during viral infection. Our findings revealed that SIRPα limits the activation and cytotoxicity of NK cells. The lack of SIRPα is associated with the downregulation of the SHP-1 molecule, which inhibits NK cell activation. By this mechanism, SIRPα prevents the proliferation and activation of NK cells. We further observed an increased NK cell-mediated killing of activated CD8+ T cells in absence of SIRPα, compromising antiviral T cell response, exacerbating LCMV virus replication and its consequences. Accordingly, the depletion of NK cells rescued the declined CD8+ T cells, alleviating the liver immunopathology and viral dissemination.
Therapeutically, our findings complement other recent approaches targeting CD47-SIRPα signaling on NK cells. Absence or blockade of SIRPα have been reported to significantly enhance NK cell activation and NK cell mediated killing of K562 tumor cells, MHC-I target cells and colon cancer [28, 29]. Notably, the upregulation of SIRPα on NK cells counters other stimulatory molecules such as NKG2D, CD16 and IL-2, therefore, inhibiting the NK cell activation signaling process [28]. Our study is in consistent with these reports as we revealed that the genetically knockout of SIRPα led to an increased NK cell activation and lysis of RMA and RMA-S target cells. Similarly, CD47−/− NK cells correspondingly displayed augmented effector phenotypes, indicating an inhibitory function of CD47 on NK cells [24]. Therapeutic antibodies that block inhibitory CD47 signaling are shown to enhance NK cell mediated tumor regression of melanomas [24] suggesting that the disruption of CD47-SIRPα signaling enhances NK cells effector function. Our data on the downregulation of SHP-1 in SIRPα deficient NK cells may in part explain the mechanism of SIRPa mediated NK cells inhibition. This is consistent with other studies linking the downregulation or knockout of SHPs to increased NK cells activation [43, 44]. SHP-1 is also known to be an essential molecule in the regulation and prevention of NK fratricide [45]. Ours and other findings on the role of SIRPα indicate that SIRPα is an inhibitory receptor inhibiting NK cell functions by recruiting and activating SHP-1, therefore, defining a threshold for NK cells activity [10-13].
In addition to NK cell antitumoral or antiviral activity, NK cells can mediated killing of CD8+ T cells [36] and can serve as rheostats for T cell response [37]. For instance, NK cells can target anti-HBV–specific T cells, contributing to T cell dysfunction [46]. Accordingly, our results demonstrate that the lack of SIRPα impaired antiviral CD8+ T cells immune responses, increased liver damage, and worsened the LCMV infection. We observed an enhanced NK cell mediated killing of endogenous and exogenous CD8+ T cells in the SIRPα deficient mice. The diametric responses might not be hardwired within SIRPα but rather result from the fact that NK cells restrict CD8+ T cells. Taking away an immunological checkpoint of NK cells causes an opposing effect on T immunity. Since CD8 T cells are crucial for LCMV control, the loss of CD8 T cells was associated with an impaired virus control in SIRPα deficient mice. However, an experimental depletion of NK cell in both SIRPα-sufficient and - deficient mice led to partial restoration of CD8 T cells and efficient virus control. It is important to understand that NK cell mediated T cell inhibition during viral infection are complex and finely tuned process, and there are many unknown factors regulating T cell immunity.
Recent clinical studies have shed new light on the role of SIRPα in cancer immunotherapy as well as its clinical relevance as prognostic marker. The importance of our findings is that we identified SIRPα as a critical inhibitory receptor that regulates NK cells activation and function. Considering the role of NK cells in the early phase of tumor regression and certain viral infections, the disruption of SIRPα signaling could be amendable as a potential target for improving NK cells function in cancer and infectious diseases. However, therapeutical targeting SIRPα need to put into account for a possible consequence of NK cell mediated killing of T cells.
Acknowledgements
This research was funded by the Deutsche Forschungsgemeinschaft (DFG) grant through the Research Training Group RTG-1949, University of Duisburg-Essen. This research was conducted within the framework of HH Sheikh Jaber Al-Ahmad Al-Sabah Chair of Microbiology & Immunology at the Arabian Gulf University.
Disclosure Statement
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors have declared that no competing interests exist.
References
| 1 | Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat
Immunol. 2008;9(5):503-10.
https://doi.org/10.1038/ni1582 |
| 2 | Hamdan TA. The Multifaceted Roles of NK Cells in the Context of Murine Cytomegalovirus and
Lymphocytic Choriomeningitis Virus Infections. Immune Netw. 2024;24(4):e29.
https://doi.org/10.4110/in.2024.24.e29 |
| 3 | Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface
receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol.
2019;16(5):430-41.
https://doi.org/10.1038/s41423-019-0206-4 |
| 4 | Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in
cancer. Nat Rev Immunol. 2018;18(11):671-88.
https://doi.org/10.1038/s41577-018-0061-z |
| 5 | Xu HC, Grusdat M, Pandyra AA, Polz R, Huang J, Sharma P, et al. Type I interferon protects
antiviral CD8+ T cells from NK cell cytotoxicity. Immunity. 2014;40(6):949-60.
https://doi.org/10.1016/j.immuni.2014.05.004 |
| 6 | Grudzien M, Rapak A. Effect of Natural Compounds on NK Cell Activation. J Immunol Res.
2018;2018:4868417.
https://doi.org/10.1155/2018/4868417 |
| 7 | Prinz D, Klein K, List J, Knab VM, Menzl I, Leidenfrost N, et al. Loss of NKG2D in murine NK cells
leads to increased perforin production upon long-term stimulation with IL-2 Eur J Immunol. 2020.
https://doi.org/10.1002/eji.201948222 |
| 8 | Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, et al. Absence of NKG2D
ligands defines leukaemia stem cells and mediates their immune evasion. Nature.
2019;572(7768):254-9.
https://doi.org/10.1038/s41586-019-1410-1 |
| 9 | Purdy AK, Campbell KS. SHP-2 expression negatively regulates NK cell function. J Immunol.
2009;183(11):7234-43.
https://doi.org/10.4049/jimmunol.0900088 |
| 10 | Viant C, Fenis A, Chicanne G, Payrastre B, Ugolini S, Vivier E. SHP-1-mediated inhibitory signals
promote responsiveness and anti-tumour functions of natural killer cells. Nat Commun. 2014;5:5108.
https://doi.org/10.1038/ncomms6108 |
| 11 | Wahle JA, Paraiso KH, Kendig RD, Lawrence HR, Chen L, Wu J, et al. Inappropriate recruitment and
activity by the Src homology region 2 domain-containing phosphatase 1 (SHP1) is responsible for
receptor dominance in the SHIP-deficient NK cell. J Immunol. 2007;179(12):8009-15.
https://doi.org/10.4049/jimmunol.179.12.8009 |
| 12 | Yusa S, Catina TL, Campbell KS. SHP-1- and phosphotyrosine-independent inhibitory signaling by a
killer cell Ig-like receptor cytoplasmic domain in human NK cells. J Immunol. 2002;168(10):5047-57.
https://doi.org/10.4049/jimmunol.168.10.5047 |
| 13 | Lowin-Kropf B, Kunz B, Beermann F, Held W. Impaired natural killing of MHC class I-deficient
targets by NK cells expressing a catalytically inactive form of SHP-1 J Immunol.
2000;165(3):1314-21.
https://doi.org/10.4049/jimmunol.165.3.1314 |
| 14 | Matozaki T, Murata Y, Okazawa H, Ohnishi H. Functions and molecular mechanisms of the
CD47-SIRPalpha signalling pathway. Trends Cell Biol. 2009;19(2):72-80.
https://doi.org/10.1016/j.tcb.2008.12.001 |
| 15 | Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal
regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc
Natl Acad Sci U S A. 2012;109(17):6662-7.
https://doi.org/10.1073/pnas.1121623109 |
| 16 | Feng Y, Huang C, Wang Y, Chen J. SIRPalpha: A key player in innate immunity. Eur J Immunol.
2023;53(11):e2350375.
https://doi.org/10.1002/eji.202350375 |
| 17 | Sosale NG, Rouhiparkouhi T, Bradshaw AM, Dimova R, Lipowsky R, Discher DE. Cell rigidity and shape
override CD47's "self"-signaling in phagocytosis by hyperactivating myosin-II. Blood.
2015;125(3):542-52.
https://doi.org/10.1182/blood-2014-06-585299 |
| 18 | Liu Y, Wang Y, Yang Y, Weng L, Wu Q, Zhang J, et al. Emerging phagocytosis checkpoints in cancer
immunotherapy. Signal Transduct Target Ther. 2023;8(1):104.
https://doi.org/10.1038/s41392-023-01365-z |
| 19 | Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, et al. Dendritic Cells but Not Macrophages Sense Tumor
Mitochondrial DNA for Cross-priming through Signal Regulatory Protein alpha Signaling. Immunity.
2017;47(2):363-73 e5.
https://doi.org/10.1016/j.immuni.2017.07.016 |
| 20 | Saito Y, Respatika D, Komori S, Washio K, Nishimura T, Kotani T, et al. SIRPalpha(+) dendritic
cells regulate homeostasis of fibroblastic reticular cells via TNF receptor ligands in the adult
spleen. Proc Natl Acad Sci U S A. 2017;114(47):E10151-E60.
https://doi.org/10.1073/pnas.1711345114 |
| 21 | Liu Q, Wen W, Tang L, Qin CJ, Lin Y, Zhang HL, et al. Inhibition of SIRPalpha in dendritic cells
potentiates potent antitumor immunity. Oncoimmunology. 2016;5(9):e1183850.
https://doi.org/10.1080/2162402X.2016.1183850 |
| 22 | Myers LM, Tal MC, Torrez Dulgeroff LB, Carmody AB, Messer RJ, Gulati G, et al. A functional subset
of CD8(+) T cells during chronic exhaustion is defined by SIRPalpha expression. Nat Commun.
2019;10(1):794.
https://doi.org/10.1038/s41467-019-08637-9 |
| 23 | Cham LB, Rosas-Umbert M, Lin L, Tolstrup M, Sogaard OS. Single-Cell Analysis Reveals That CD47
mRNA Expression Correlates with Immune Cell Activation, Antiviral Isgs, and Cytotoxicity. Cell
Physiol Biochem. 2024;58(4):322-35.
https://doi.org/10.33594/000000715 |
| 24 | Nath PR, Pal-Nath D, Mandal A, Cam MC, Schwartz AL, Roberts DD. Natural Killer Cell Recruitment
and Activation Are Regulated by CD47 Expression in the Tumor Microenvironment. Cancer Immunol Res.
2019;7(9):1547-61.
https://doi.org/10.1158/2326-6066.CIR-18-0367 |
| 25 | Nath PR, Gangaplara A, Pal-Nath D, Mandal A, Maric D, Sipes JM, et al. CD47 Expression in Natural
Killer Cells Regulates Homeostasis and Modulates Immune Response to Lymphocytic Choriomeningitis
Virus. Front Immunol. 2018;9:2985.
https://doi.org/10.3389/fimmu.2018.02985 |
| 26 | Kaur S, Bronson SM, Pal-Nath D, Miller TW, Soto-Pantoja DR, Roberts DD. Functions of
Thrombospondin-1 in the Tumor Microenvironment. Int J Mol Sci. 2021;22(9).
https://doi.org/10.3390/ijms22094570 |
| 27 | Lang B, Wang M, Zhang Z, Fu Y, Han X, Hu Q, et al. Inhibitory receptor CD47 binding to plasma TSP1
suppresses NK-cell IFN-gamma production via activating the JAK/STAT3 pathway during HIV infection. J
Transl Med. 2023;21(1):869.
https://doi.org/10.1186/s12967-023-04667-6 |
| 28 | Deuse T, Hu X, Agbor-Enoh S, Jang MK, Alawi M, Saygi C, et al. The SIRPalpha-CD47 immune
checkpoint in NK cells. J Exp Med. 2021;218(3).
https://doi.org/10.1084/jem.20200839 |
| 29 | Yilmaz L, Oztuzcu S, Eronat O, Ugur BK, Gumus M, Guler S, et al. Exploring NK cell dynamics and
the CD47-SIRPalpha axis in colon cancer. Hum Immunol. 2025;86(2):111254.
https://doi.org/10.1016/j.humimm.2025.111254 |
| 30 | Cruz Tleugabulova M, Zhao M, Lau I, Kuypers M, Wirianto C, Umana JM, et al. The Protein
Phosphatase Shp1 Regulates Invariant NKT Cell Effector Differentiation Independently of TCR and Slam
Signaling. J Immunol. 2019;202(8):2276-86.
https://doi.org/10.4049/jimmunol.1800844 |
| 31 | Niogret C, Miah SMS, Rota G, Fonta NP, Wang H, Held W, et al. Shp-2 is critical for ERK and
metabolic engagement downstream of IL-15 receptor in NK cells. Nat Commun. 2019;10(1):1444.
https://doi.org/10.1038/s41467-019-09431-3 |
| 32 | Huang A, Shinde PV, Huang J, Senff T, Xu HC, Margotta C, et al. Progranulin prevents regulatory NK
cell cytotoxicity against antiviral T cells. JCI Insight. 2019;4(17).
https://doi.org/10.1172/jci.insight.129856 |
| 33 | Khairnar V, Duhan V, Patil AM, Zhou F, Bhat H, Thoens C, et al. CEACAM1 promotes CD8(+) T cell
responses and improves control of a chronic viral infection. Nat Commun. 2018;9(1):2561.
https://doi.org/10.1038/s41467-018-04832-2 |
| 34 | Hamdan TA, Ashraf F, Bhat H. Insights into Virus-Induced Immune Mediated Liver Pathology. Cell
Physiol Biochem. 2022;56(4):340-52.
https://doi.org/10.33594/000000554 |
| 35 | Benson RA, MacLeod MK, Hale BG, Patakas A, Garside P, Brewer JM. Antigen presentation kinetics
control T cell/dendritic cell interactions and follicular helper T cell generation in vivo. Elife.
2015;4.
https://doi.org/10.7554/eLife.06994 |
| 36 | Cook KD, Waggoner SN, Whitmire JK. NK cells and their ability to modulate T cells during virus
infections. Crit Rev Immunol. 2014;34(5):359-88.
https://doi.org/10.1615/CritRevImmunol.2014010604 |
| 37 | Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating
antiviral T cells. Nature. 2011;481(7381):394-8.
https://doi.org/10.1038/nature10624 |
| 38 | Lang PA, Lang KS, Xu HC, Grusdat M, Parish IA, Recher M, et al. Natural killer cell activation
enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc Natl
Acad Sci U S A. 2012;109(4):1210-5.
https://doi.org/10.1073/pnas.1118834109 |
| 39 | Pallmer K, Barnstorf I, Baumann NS, Borsa M, Jonjic S, Oxenius A. NK cells negatively regulate CD8
T cells via natural cytotoxicity receptor (NCR) 1 during LCMV infection. PLoS Pathog.
2019;15(4):e1007725.
https://doi.org/10.1371/journal.ppat.1007725 |
| 40 | Duhan V, Hamdan TA, Xu HC, Shinde P, Bhat H, Li F, et al. NK cell-intrinsic FcepsilonRIgamma
limits CD8+ T-cell expansion and thereby turns an acute into a chronic viral infection. PLoS Pathog.
2019;15(6):e1007797.
https://doi.org/10.1371/journal.ppat.1007797 |
| 41 | Veillette A, Chen J. SIRPalpha-CD47 Immune Checkpoint Blockade in Anticancer Therapy. Trends
Immunol. 2018;39(3):173-84.
https://doi.org/10.1016/j.it.2017.12.005 |
| 42 | Ma CS, Nichols KE, Tangye SG. Regulation of cellular and humoral immune responses by the SLAM and
SAP families of molecules. Annu Rev Immunol. 2007;25:337-79.
https://doi.org/10.1146/annurev.immunol.25.022106.141651 |
| 43 | Wu N, Zhong MC, Roncagalli R, Perez-Quintero LA, Guo H, Zhang Z, et al. A hematopoietic
cell-driven mechanism involving SLAMF6 receptor, SAP adaptors and SHP-1 phosphatase regulates NK
cell education. Nat Immunol. 2016;17(4):387-96.
https://doi.org/10.1038/ni.3369 |
| 44 | Matalon O, Ben-Shmuel A, Kivelevitz J, Sabag B, Fried S, Joseph N, et al. Actin retrograde flow
controls natural killer cell response by regulating the conformation state of SHP-1 EMBO J.
2018;37(5).
https://doi.org/10.15252/embj.201696264 |
| 45 | Mahmood S, Kanwar N, Tran J, Zhang ML, Kung SK. SHP-1 phosphatase is a critical regulator in
preventing natural killer cell self-killing. PLoS One. 2012;7(8):e44244.
https://doi.org/10.1371/journal.pone.0044244 |
| 46 | Schuch A, Hoh A, Thimme R. The role of natural killer cells and CD8(+) T cells in hepatitis B
virus infection. Front Immunol. 2014;5:258.
https://doi.org/10.3389/fimmu.2014.00258 |