Review Article – DOI: 10.33594/000000864
CPB (60): 223 - 244
Accepted: 15.01.2026 - Published: 29.04.2026
Macrophage Migration Inhibitory Factor and Pulmonary Immunity: a Systems Biology Perspective on Its Role in Lung Diseases
bDepartment of Public Health and Healthcare Organization, Azerbaijan Medical University, Baku, Azerbaijan ,
cDepartment of Pharmaceutical Toxicology and Chemistry, Azerbaijan Medical University, Baku, Azerbaijan,
dDepartment of Human Anatomy and Medical Terminology, Azerbaijan Medical University, Baku, Azerbaijan
Keywords
Abstract
Macrophage migration inhibitory factor (MIF) is a pleiotropic cytokine that occupies a central regulatory position within pulmonary immune networks, integrating inflammatory signalling, redox control, and immune stromal communication. Originally characterised as a pro-inflammatory mediator, MIF is now recognised to exert context-dependent functions that range from: protective host defence during acute infection to the promotion of chronic inflammation, fibrosis, and tumour progression when regulatory constraints fail. This review synthesises current evidence on the molecular biology, structural features, and signalling mechanisms governing MIF activity in the lung, emphasising its role as a network hub coordinating CD74/CD44- and CXCR-mediated signalling, glucocorticoid antagonism, and redox imbalance. A systems biology perspective is applied to illustrate how genetic variability, environmental exposure, ageing, and metabolic stress reprogram MIF-centred immune circuits across pulmonary disease states. Integration of multi-omics insights, systems pharmacology, and computational modelling highlights emerging opportunities for the selective modulation of MIF signalling, rather than indiscriminate inhibition. Disease-specific manifestations of MIF dysregulation in pneumonia, chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis, and lung cancer are discussed as dynamic outcomes of shared regulatory architectures. Collectively, this review positions MIF as a critical immunoregulatory node whose context-dependent modulation may support future biomarker development and therapeutic strategies currently under active preclinical investigation in complex pulmonary disorders.
Introduction
Chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF), acute respiratory distress syndrome (ARDS), and lung cancer are still among the leading causes of morbidity and mortality in the world especially in the ageing countries. Aging is linked to an increasingly impaired balance of immune homo-ostasis and development of chronic low-grade inflammation, often known as inflammaging. The chronic inflammatory condition conditions an individual to exaggerated immune responses to acute insults in addition to crippling effective tissue repair processes. As a result, impairments of immune homeostasis lead to pathological outcomes such as structural remodelling, fibrosis and malignant change of pulmonary tissues. Notably, such pathological processes are hardly due to the malfunction of one molecular pathway. Rather, they are indicative of intricate perturbations of cytokine centred signalling networks which mediate inflammation, oxidative stress and cell survival. Growing evidence indicates that this development of pulmonary diseases is caused by a lack of functionality of integrated immunoregulatory circuits, and is not due to solitary signalling defects. These interrelated regulatory systems are thus very important in explaining disease progression and determining the rational therapeutic targets [1]. Macrophage migration inhibitory factor (MIF) has become a key controller of these networks. MIF was identified in the first place as an agent able to suppress the migration of macrophages, but it is today also regarded as a pleiotropic cytokine that integrates innate and adaptive immune responses, metabolic adaptation, and cellular stress signalling [2]. This is due to its functional diversity occasioned by its interactions with several receptor systems such as the CD74/CD44 complex and CXCR2, CXCR4 and CXCR7 chemokine receptors. These receptor interactions allow MIF to connect MAPK/ERK and PI3K/Akt signalling with NF-kB activation and redox-sensitive transcriptional programs and, thus, to integrate inflammatory signalling with cellular proliferation, migration and survival [3]. In addition to its role as a canonical inflammatory mediator, MIF is also an immune activation/redox interface. It helps to induce glucocorticoid resistance, regulate macrophage activation conditions and has intrinsic tautomerase and oxidoreductase functions that affect oxidative balance [4]. Persistent MIF expression or uncontrolled MIF expression has been linked with continuous cytokine expression, escalation of oxidative stress and dysadaptive tissue remodelling in chronic pulmonary diseases. It has also been shown by use of neuroinflammatory and systemic inflammatory models that MIF signalling is extremely context-dependent, differing depending on cellular environment, regulatory feedback mechanisms and signal duration [5, 6]. These observations indicate that the intensity of signalling and the dynamics of signalling determine whether MIF responses are sustaining or progress to pathogenic responses. Compared to the previous review in Cellular Physiology and Biochemistry (2025), which analyzed the larger role of the macrophage migration inhibitory factor in a variety of organ systems [1], the current manuscript is specifically on pulmonary immunity and lung disease. Here, MIF is conceptualised as a context-sensitive regulatory hub that influences the balance between protective host defence and pathogenic tissue remodelling within the lung. By integrating receptor-mediated signalling, glucocorticoid responsiveness, and redox regulation within pulmonary immune networks, this review explores how MIF shapes disease phenotypes in COPD, ARDS, idiopathic pulmonary fibrosis, and lung cancer. Applying a systems biology perspective, the manuscript further discusses emerging translational insights and potential therapeutic strategies targeting MIF signalling within pulmonary disease contexts.
Literature Search Strategy
A literature search was conducted in Google Scholar, Science Direct, PubMed and DOAJ covering publications from 2000 to 2026. Search terms included “macrophage migration inhibitory factor,” “pulmonary immunity,” “COPD,” “idiopathic pulmonary fibrosis,” “ARDS,” “lung cancer,” “glucocorticoid resistance,” “redox signalling,” “CXCR4,” and “CD74.” Studies providing mechanistic, translational, or clinical insights into MIF signalling in pulmonary contexts were prioritised. Non-respiratory studies or articles lacking mechanistic relevance were excluded. Particular attention was given to studies describing receptor interactions, downstream signalling cascades, feedback regulation, and disease stage-specific effects.
Molecular Biology and Structural Basis of MIF
Gene organisation and polymorphisms
Chromosome 22q11.2 carries the MIF gene, which encodes a 12.5 kDa protein that is highly conserved. Transcriptional activity and circulating MIF are controlled by promoter polymorphisms, with the tetranucleotide CATT repeat and the -173 G/C single-nucleotide polymorphism being the most significant ones, which influence inter-individual variability in immune responsiveness [7-9]. CATT7–8 repeats and the −173 C allele are associated with higher MIF transcription and are linked to hyper-inflammatory phenotypes, glucocorticoid resistance, and reduced immune resolution [9, 10]. Persistent MIF stimulation through such variants strengthens macrophage activation and redox imbalance, which favour chronic inflammatory and fibrotic pulmonary diseases, such as COPD, asthma, and idiopathic pulmonary fibrosis [11]. These genetic variants are particularly relevant in pulmonary inflammatory disorders, where increased MIF expression may amplify immune activation, oxidative stress, and tissue remodelling within the lung microenvironment [12-14]. Table 1 summarises important MIF variant genotype alterations and their association with diseases.
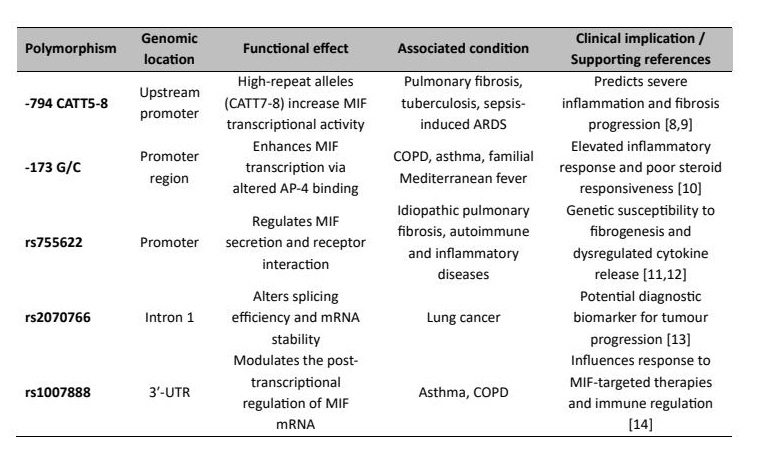
Table 1: Key MIF gene variants and their functional links to respiratory disease susceptibility
Protein structure and catalytic motifs
MIF is a homotrimeric protein where each monomer is involved in a central catalytic pocket that contains tautomerase and oxidoreductase activity [15]. The thiol-disulfide exchange and redox-sensitive immune modulation are regulated by catalytic residues, such as Pro 1 and Lys 32. The interactions with CD74/CD44 and chemokine receptors, CXCR2, CXCR4, and CXCR7, are also regulated by this trimeric structure, which connects the molecular conformation with the immune cell recruitment and survival signals [16]. Fig. 1 shows these structure-function relationships.
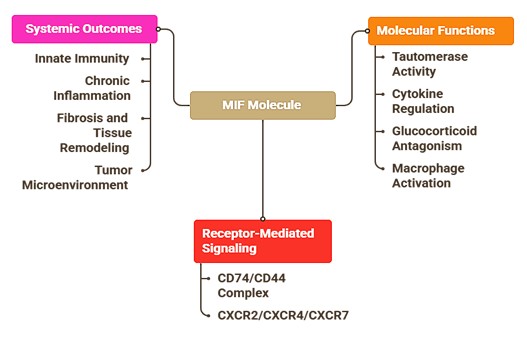
Fig. 1: Overview of the macrophage migration inhibitory factor (MIF) molecule and its systemic effects.
Advances in inhibitor design and structural modulation
Medicinal chemistry initiatives have led to the identification of MIF antagonists that include ISO-1, 4-IPP or benzoxazole or isoxazole analogues, which covalently bind to the tautomerase active site and inhibit downstream inflammatory and fibrotic signalling [17]. MIF also interacts with CD74/CD44, which is blocked by these inhibitors, and this also restores corticosteroid sensitivity in refractory airway inflammation. Optimisation approaches focus on the use of inhalable and nanoparticle delivery products to localise the reestablishment of MIF-directed immune networks to pulmonary bioavailability, metabolic stability, and tissue selectivity to minimise systemic effects.
Cellular sources and regulation of expression
Macrophages, T lymphocytes, airway epithelial cells, and endothelial cells constitutively express MIF that is rapidly released upon infection, oxidative stress or hypoxia [18]. Lack of classical signal peptide allows timely secretion and redistribution within the microenvironment of the alveolus. In pulmonary tissues, MIF expression in alveolar macrophages and airway epithelial cells enables rapid amplification of inflammatory responses during infection, environmental stress, or tissue injury. The environmental exposures, such as the cigarettes smoke and ozone, also increase MIF expression and reinforce immune activation. In as much as this facilitates host defence, persistent overexpression stabilises maladaptive inflammatory states that facilitate tissue remodelling and tissue loss [19].
Clinical relevance of MIF expression in pulmonary disorders
High levels of circulating and alveolar MIF are associated with the severity of the disease in COPD and idiopathic pulmonary fibrosis, which is an indication of persistent immune activation and regulatory imbalance [20]. MIF facilitates the recruitment of macrophages and neutrophils in COPD and fibroblast activation and the deposition of extracellular matrix in fibrotic disease. Within the lung microenvironment, MIF signalling also influences interactions among immune cells, airway epithelial cells, and pulmonary fibroblasts, thereby contributing to inflammatory amplification and structural remodelling. These trends indicate that MIF expression is a symptom of dysregulation of systemic immunity, rather than an aggregate outcome of local inflammation, and so MIF is a biomarker and a treatment target that requires lung- selective pharmacokinetics optimised therapy [21].
Signalling Pathways and Molecular Mechanisms
MIF receptor systems: CD74/CD44 complex and chemokine-like signalling
Macrophage migration inhibitory factor (MIF) has its effects on the cells by a variety of receptor systems, the most eminent of which is the CD74/CD44. CD74 is the main receptor that helps the recruitment of CD44 after MIF binding and subsequent downstream MAPK/ERK and PI3K/Akt signalling pathways that mediate cell survival, proliferation, and cytokine production [22]. This signalling cascade links inflammatory signal transduction with structural cellular responses in pulmonary tissues. The activation of ERK1/2 and focal adhesion kinase (FAK) in endothelial and smooth muscle cells facilitates cellular proliferation, migration, and alterations in vascular tone, which are part of vascular remodelling and pulmonary hypertension [23]. Simultaneously, MIF acts as a non-cognate ligand of chemokine receptors CXCR2, CXCR4, and CXCR7, which promotes leukocyte chemotaxis, neutrophil recruitment, and monocyte adhesion. Cytokine and chemokine signalling facilitates communication between immune and structural compartments to promote airway remodelling, vascular inflammation, and fibrotic pathways in chronic lung disease [24].
Crosstalk with glucocorticoid signalling
Another characteristic of MIF biology is its antagonistic action toward glucocorticoid signalling. Unlike many cytokines, MIF expression is induced by glucocorticoids and subsequently counteracts the anti-inflammatory effects of glucocorticoids, maintaining inflammation despite corticosteroid treatment [25]. MIF suppresses MAPK phosphatase-1 (MKP-1) and thus maintains p38 and ERK activation, thereby prolonging the production of cytokines [22]. MIF also disrupts glucocorticoid receptor nuclear translocation and DNA binding, further inhibiting glucocorticoid-mediated repression of genes. MIF-mediated glucocorticoid resistance is associated with sustained macrophage activation, increased oxidative stress, and reduced therapeutic responsiveness in severe asthma, COPD, and ARDS [26]. These mechanisms suggest that MIF signalling may represent a potential therapeutic target for overcoming glucocorticoid resistance in treatment-refractory pulmonary inflammation.
Regulation of oxidative and redox pathways
Oxidative stress is one of the typical features of chronic pulmonary inflammation, and MIF plays an important role in regulating cellular redox balance. MIF is involved in the control of thiol–disulfide exchange and reactive oxygen species (ROS) through intrinsic oxidoreductase activity mediated by conserved cysteine and proline residues under physiological conditions [27]. However, sustained MIF expression shifts cellular redox balance toward oxidative stress, which enhances the activity of NADPH oxidase and promotes mitochondrial dysfunction. MIF enhances NF-κB activation, increasing expression of pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β, while concurrently suppressing Nrf2-mediated antioxidant responses [28, 29]. Such effects are also compounded by environmental stressors, including cigarette smoke, ozone, and particulate pollutants, which increase the expression of MIF and strengthen a feed-forward loop between the oxidative stress and inflammation [30]. Importantly, these signalling pathways do not operate independently but form an interconnected regulatory network. Activation of the CD74/CD44 receptor complex by MIF initiates MAPK/ERK and PI3K/Akt signalling, which enhances NF-κB-dependent transcription of pro-inflammatory cytokines [22]. Simultaneously, sustained MIF signalling suppresses Nrf2-mediated antioxidant responses, promoting oxidative stress and mitochondrial dysfunction. The resulting redox imbalance further amplifies NF-κB activation, generating a feed-forward inflammatory loop [27]. Simultaneously, MIF disrupts glucocorticoid receptor signalling by inhibiting MAPK phosphatase-1 and blocking nuclear translocation of receptor, and therefore inflammatory signalling despite corticosteroid treatment. This signalling circuit offers a mechanistic description of chronic and incurable pulmonary diseases like COPD and ARDS which have persistent inflammation and steroid resistance [19].
Downstream immune modulation and macrophage polarisation
MIF has a wide range of downstream effects on immune regulation and one of the most important effector pathways is polarisation of macrophages. MIF promotes a pro-inflammatory M1 phenotype, which enhances the production of nitric oxide, release of cytokines and antigen presentation, which involves the pathogenesis of acute and chronic lung disease [31]. Simultaneously, MIF suppresses M2 polarisation by blocking the effect of the STAT6- and IL-10-dependent signalling, consequently increasing tissue repair and resolve. MIF also triggers T cell activation and neutrophil recruitment via CXCR2 and CXCR4, resulting in an inflammatory network of epithelial cells, macrophages and neutrophils tightly connected with each other. These interactions preserve immune activation and cause progressive tissue remodelling in chronic pulmonary diseases like COPD, asthma, and pulmonary fibrosis [32]. The important MIF-regulated signalling pathways and functional consequences are listed in Table 2. Importantly, the biological impact of these pathways depends not only on their activation, but also on the magnitude and duration of MIF signalling.
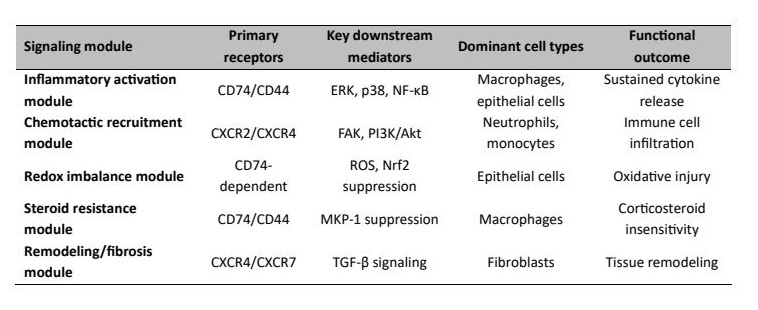
Table 2: MIF-Regulated Signalling Modules and Functional Outcomes in Pulmonary Immunity
Signal Intensity and Temporal Dynamics of MIF Activation
Macrophage migration inhibitory factor (MIF) has biological effects that are dependent on the intensity and duration of signalling. In acute infection, transient MIF increase promotes macrophage activation and leukocyte recruitment, facilitating coordinated inflammatory responses and efficient host defence [2, 33]. In that case, immune activation remains self-limiting and is followed by effective tissue repair. Conversely, sustained high-amplitude MIF expression promotes persistent inflammatory signalling and glucocorticoid resistance, maintaining immune activation beyond the period required for host defence [2]. The long-term sustained increase of MIF in pulmonary tissues, such as that observed in COPD, strengthens macrophage-mediated inflammation and contributes to progressive structural remodelling [19]. This supports a threshold-sensitive model in which regulated MIF signalling maintains protective immunity, whereas sustained dysregulation alters pulmonary immune networks toward chronic inflammation and disease progression. These observations support a threshold-dependent framework in which MIF functions as a regulatory hub whose biological effects depend on signalling amplitude, duration, and tissue context. The integrative systems model of the MIF-centred pulmonary immune response is described in Fig. 2, in which MIF acts as the central regulatory node connecting receptor-mediated signalling (CD74/CD44, CXCR2/4/7), NF-κB signalling, and redox regulation. Downstream immune outcomes, including COPD, IPF, ARDS, and lung cancer, are determined by contextual modifiers such as genetic polymorphisms, ageing, environmental stress, and metabolic factors.
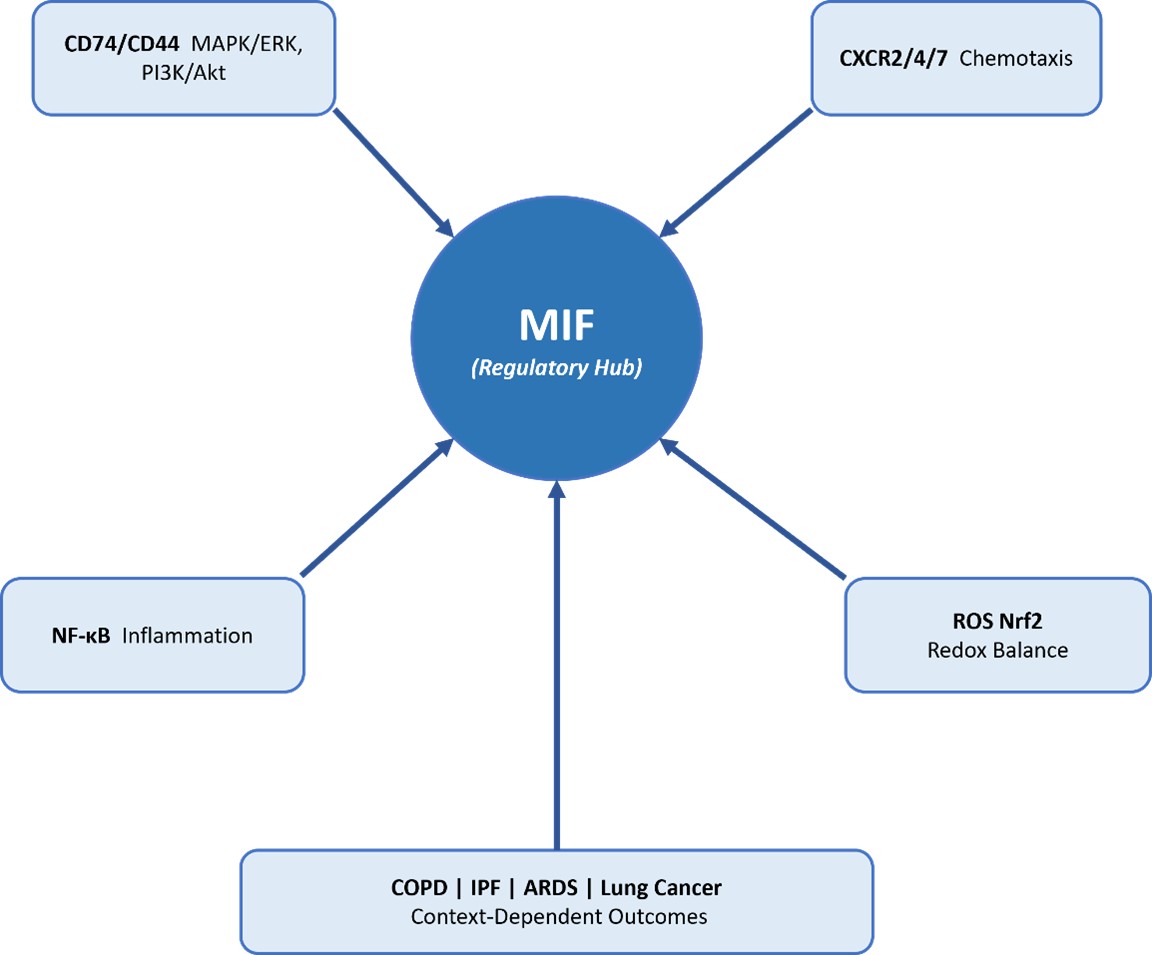
Fig. 2: Context-Dependent Systems Model of MIF-Centred Pulmonary Immune Regulation.
Integration of Public Omics and Systems-Level Data
Publicly available transcriptomic and proteomic datasets further support the role of macrophage migration inhibitory factor (MIF) in pulmonary disease. Gene expression data from the Gene Expression Omnibus (GEO), including datasets such as GSE47460 (COPD) and GSE70866 (idiopathic pulmonary fibrosis), indicate increased MIF expression in diseased lung tissue and immune cell populations. In addition, datasets related to acute lung injury (e.g., GSE76293) suggest upregulation of MIF during early inflammatory responses. Protein-level data from the Human Protein Atlas demonstrate MIF expression in alveolar macrophages and bronchial epithelial cells, consistent with its role in pulmonary immune regulation. Furthermore, protein–protein interaction analyses using the STRING database highlight functional associations of MIF with CD74, CXCR4, and key inflammatory mediators, supporting its role as a central regulatory hub. Together, these publicly available datasets reinforce the concept of MIF as a context-dependent modulator of pulmonary immune responses across multiple disease states. To further support the mechanistic framework, publicly available transcriptomic, proteomic, and systems biology datasets provide independent evidence for the role of macrophage migration inhibitory factor (MIF) in pulmonary diseases (Table 3).
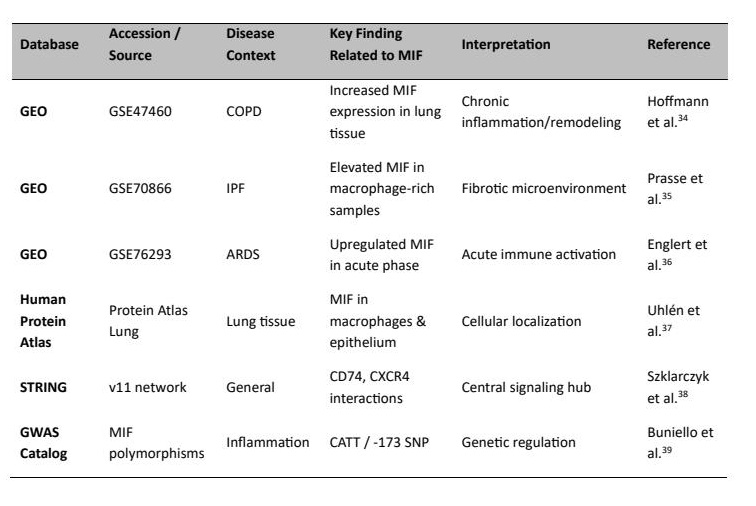
Table 3: Publicly Available Omics Datasets Supporting the Role of MIF in Pulmonary Diseases
MIF in Pulmonary Immunity and Host Defence
Role in innate immune responses
The main role of Macrophage migration inhibitory factor (MIF) is in the regulation of macrophage activation, phagocytosis and the production of inflammatory mediators through the coordination of these processes. MIF in the pulmonary environment increases early host responses to infection by increasing pathogen recognition and microbial killing. In intracellular infection like Mycobacterium tuberculosis, MIF is rapidly induced in alveolar macrophages to help in the successful clearance of the bacteria, and this has been demonstrated to be essential in pulmonary host defence [33]. MIF increases phagocytosis by increasing scavenger receptor and Toll-like receptor signalling, specifically TLR2 and TLR4, to maximise the elimination of pathogens. In addition to macrophages, MIF controls neutrophil recruitment by CXCR2-and CXCR4-mediated chemotactic pathways, which mediate the immune cell trafficking to inflamed alveolar compartments [40]. Even though it is necessary to control early microbial invasion, excessive neutrophilic infiltration is a contributor to tissue injury. Neutrophil accumulation driven by MIF has been implicated in acute respiratory distress syndrome (ARDS) and ventilator-induced lung injury, in which excessive release of cytokines disrupts the integrity of the alveolar barrier and increases the permeability of the vasculature [41]. By regulating upstream cytokines such as TNF-α, IL-6, and IL-8, MIF coordinates inflammatory amplification loops during acute lung inflammation. MIF also balances pro- and anti-inflammatory mediators and regulates IL-10 and IL-1 receptor antagonist signalling to control the intensity of the immune response. The controlled activation is beneficial to defence, whereas the dysregulated signalling is facilitative of the development of cytokine storm and pathological inflammation [42]. MIF is an important mediator between innate immune surveillance and adaptive immune interaction in pulmonary infections through these mechanisms.
Adaptive immune modulation
MIF has a wide regulatory effect on adaptive immunity that affects T-cell differentiation, antigen presentation, and immune tolerance. It promotes Th1 polarisation through IL-12 and IFN-γ signalling while suppressing Th2-associated cytokines such as IL-4 and IL-10, thereby enhancing antimicrobial defence [43]. Nevertheless, chronic Th1-biased responses are associated with long-term pulmonary disease and inflammatory damage. Simultaneously, MIF boosts IL-6/IL-23 signalling, which promotes Th17 differentiation and supports the process of proinflammatory cytokine cascades, which create a Th1/Th17-biased immune response in asthma, COPD, and interstitial lung disease. MIF also improves the antigen-presenting cell activity by increasing the MHC class II-molecules and costimulatory markers CD80 and CD86 on dendritic cells and macrophages, and stimulates T-cell priming and clonal expansion. Continuous MIF signalling, in its turn, impairs immune tolerance and preconditions the T-cell activation and autoimmune-like responses in pulmonary tissues over the long term [44]. MIF plays a role in immunomodulatory functions in lung cancer through the polarisation of tumour-associated macrophages to immunosuppressive phenotypes and the recruitment of myeloid-derived suppressor cells by the MIF-CD74 axis. These pathways repress cytotoxic T cells and decrease sensitivity to immune checkpoint inhibition, and increase fibrotic remodelling in patients with coexisting interstitial lung disease and lung cancer [45]. These immune interactions also influence fibroblast activation and extracellular matrix deposition, linking immune dysregulation with structural remodelling in pulmonary tissues.
MIF and the ageing lung immune environment
As age progresses, MIF is closely linked to immunosenescence and disturbed cytokine interactions in the lung. Older tissues are characterised by chronic low-grade inflammation, which is characterised by high levels of proinflammatory cytokines and reduced regenerative potential. MIF maintains the state of macrophage activation and delays inflammatory reaction, and age-related hypersensitivity of the alveolar macrophages facilitates the uncontrolled generation of reactive oxygen species and release of inflammatory mediators, accelerating the damage to the epithelial tissue and matrix deposition [46]. Persistent high levels of MIF in older patients are associated with the up-regulation of fibrotic genes, such as collagen I and fibronectin. Constant stimulation of MIF-CD74 and MIF-CXCR4 receptors promotes fibroblast growth and myofibroblast differentiation and stimulates fibrotic progression [47]. Additional age-related immune mechanisms encompass depletion of naïve T cells and accumulation of senescent T cells that express exhaustion markers (PD-1, TIM-3, etc.) that prevent immune vigilance and adaptive responsiveness. MIF-driven persistence of fibrogenic mediators, including TGF-β and IL-13, reinforces tissue stiffening and disease progression [48]. MIF or CD74 pharmacological inhibition has shown the ability to decrease fibroblast and collagen deposition activities and to at least partially restore pulmonary function [49]. In general, MIF is a protective and disruptive regulator of lifespan pulmonary immune balance. Although necessary in early defence and repair, chronic MIF signalling in ageing enhances chronic inflammation, immune fatigue and fibrotic remodelling [50]. MIF is a key intermediary between ageing, immunity and chronic lung pathology by coordinated modulation of macrophage responses, T-cell responses and immune and stromal cell interactions.
MIF in Specific Pulmonary Diseases
The manifestations of MIF dysregulation in pulmonary diseases can be interpreted through the lens of the signal intensity and temporal dynamics described above. Excessive short-term activation of MIF signalling is associated with acute conditions such as pneumonia, whereas chronic diseases such as COPD, idiopathic pulmonary fibrosis (IPF), and lung cancer are characterised by sustained MIF activation that disrupts immune homeostasis and promotes structural remodelling [28, 33]. These disease-specific outcomes arise from the signalling mechanisms described previously. Rather than acting as a disease-specific cytokine, MIF functions as a central immunoregulatory node that integrates inflammatory signalling, oxidative stress, and immune modulation across multiple pulmonary disorders [22, 29].
Pneumonia and Acute Respiratory Distress Syndrome (ARDS)
MIF is also expressed within the alveolar macrophages and endothelial cells at a rapid rate, which enhances cytokine cascades and leukocyte recruitment during acute pulmonary infection. Persistent MIF signalling increases NF-κB activation and breaks epithelial-endothelium barrier integrity, leading to a rise in vascular permeability, alveolar oedema, and the development of cytokine storms. Inhibitory activity of MIF prevents the severity of inflammation and lung permeability, which highlights its effects on immune overreaction during pneumonia and ARDS [51]. Early inflammation dysregulation may also predispose subsequent fibrotic remodelling by targeting fibroblast activation and collagen in the initial injury models induced by bleomycin blockage of MIF, where MIF inhibition prevents later fibrotic remodelling [52]. Fine-tuning of pulmonary MIF could hence alleviate acute damage, yet spare antimicrobial immunity.
Chronic Obstructive Pulmonary Disease (COPD)
The sustained MIF overexpression of airway macrophages and epithelial cells in COPD strengthens the oxidative stress and resistance to glucocorticoids. MAPK and PI3K/Akt signalling pathways are activated to promote the survival of macrophages and long-term recruitment of neutrophils to keep chronic inflammation [53]. Inter-individual differences in inflammatory burden and therapeutic response are partly influenced by genetic variants that increase MIF transcription. High MIF activity is associated with low corticosteroid effect, and pharmacological blockage of MIF restores steroid sensitivity and minimises oxidative damage [54]. The lung-targeted delivery strategies can be optimised even further to enhance long-term disease control.
Idiopathic Pulmonary Fibrosis (IPF)
MIF mediates the interaction between immune activation and fibroblast proliferation in the fibrotic lung disease, leading to progressive tissue remodelling. MIF disrupts alveolar repair by promoting the differentiation of myofibroblasts and epithelial to mesenchymal transition through ERK and focal adhesion kinase signalling [55]. Synthesis of extracellular matrix is enhanced by synergistic effects with profibrotic mediators, including transforming growth factor-β (TGF-β). MIF inhibitors and regulatory microRNAs delivered with nanocarriers offer enhanced lung retention and biodistribution, which is significant because of the necessity to regulate immune-stromal interaction in reducing fibrosis without impairing immune capability [56].
Lung Cancer
In the microenvironment of the pulmonary tumour, MIF enhances angiogenesis, immune evasion, and tumour progression by maintaining ERK and PI3K/Akt signalling. High MIF expression promotes the recruitment and polarisation of tumour-associated macrophages into an immunosuppressive phenotype and suppresses the activity of cytotoxic T cells, promoting tumour growth and metastasis. MIF pharmacological inhibition has been shown to decrease the tumour burden and the metastatic potential in experimental models [57]. The combination of MIF antagonists and immune checkpoint inhibitors, with the help of combinatorial strategies, can be effective in improving antitumor immunity, as long as sufficient pulmonary distribution and metabolic stability are attained [58]. In a wide range of pulmonary disorders, MIF occurs as a network centre that combines the signalling of cytokines, oxidative imbalance, and immune tolerance. Depending on the context of modulation of MIF activity, the immune responses can be maintained as protective or go to chronic inflammation, fibrosis, or immune escape. Therapeutic targeting of MIF may therefore represent a promising strategy for restoring immune balance and improving responsiveness to existing treatments while minimising systemic toxicity.
Dual Role of MIF: Protective vs. Pathogenic
Context-Dependent Effects of MIF
Macrophage migration inhibitory factor (MIF) is functionally dual in that it is a protective mediator in acute infection or tissue injury by enhancing leukocyte recruitment, macrophage activation and pathogen clearance, enhancing immune containment and tissue repair. On the contrary, chronic or maladaptive MIF expression maintains immune and redox signalling pathways in a persistently activated condition, which sustains long-lasting inflammation, fibrosis and tissue damage. It has been experimentally demonstrated that pharmacological inhibition of MIF can suppress pathological signalling without impairing key immune functions, which is a context-specific regulation mechanism [59]. This dual aspect places MIF as one of the major determinants of pulmonary immune homeostasis, with accurate timing control determining recovery and disease persistence.
Genetic and environmental modifiers
Biological effects of MIF highly depend on genetic variation and exposure to the environment. MIF transcription Promoter polymorphisms may alter the activity of MIF transcription that controls predisposition to inflammatory and autoimmune pulmonary diseases and contributes to inter-individual variation in immune responsiveness. MIF expression, amplification of oxidative stress, and redox imbalance are also promoted by environmental stressors like cigarette smoke and air pollution. These effects may be reinforced by exosomal exchange of MIF-regulatory microRNAs and proteins between immune and epithelial cells, to spread coordinated but maladaptive pulmonary compartmental signalling [59]. Genetic and environmental factors, together, influence the responses of MIF to be protective or pathogenic.
Dose- and duration-dependent effects
MIF activity is critically regulated by concentration and exposure duration. Transient, low-level activation supports cytoprotective mechanisms, preserves redox balance, and facilitates controlled immune responses. Maintenance of physiological redox homeostasis, including adequate micronutrient status, may support antioxidant defences and reduce amplification of oxidative inflammatory signalling [60]. In contrast, sustained overexpression promotes persistent cytokine signalling, fibroblast activation, and progressive tissue remodelling, thereby reinforcing inflammatory and fibrotic circuits [28]. Nigella sativa derivatives have been suggested as possible modulators of the inflammatory signalling pathways and oxidative stress responses through the adjunct antioxidant and anti-inflammatory bioactive compounds [61]. These findings are consistent with the threshold-dependent framework above. Transient signalling facilitates antimicrobial immunity and tissue repair, and chronic high-amplitude activation alters pulmonary immune networks to the path of chronic inflammation and structural remodelling. Thus, the biological impact of MIF depends on the balance between transient protective signalling required for host defence and sustained high-amplitude activation that drives chronic inflammation and tissue remodelling. Selective inhibition of pathogenic MIF amplification while preserving its protective immune functions should therefore be considered in therapeutic strategies (Table 4).
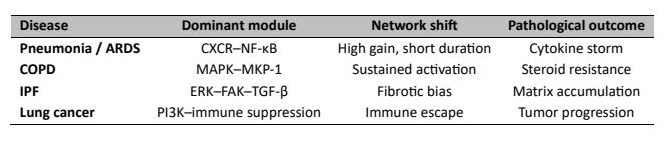
Table 4: Disease-Specific Parameter Shifts in the MIF Network
Clinical and Translational Perspectives
MIF as a biomarker of disease activity
Macrophage migration inhibitory factor (MIF) has become an attractive biomarker of pulmonary and systemic inflammatory diseases. Elevated levels of MIF in serum, bronchoalveolar lavage fluid, and exhaled breath condensates have been associated with disease activity and progression in chronic obstructive pulmonary disease (COPD), pulmonary fibrosis, and certain pulmonary malignancies. Rather than reflecting isolated inflammatory events, sustained MIF expression may indicate persistent dysregulation of immune and redox control systems within pulmonary tissues. MIF has also been identified as having altered cytokine signatures in inflammatory disorders as the possible predictor of disease severity and systemic, inflammatory, involvement [62]. Furthermore, it has been reported that MIF-enriched circulating extracellular vesicles are markers of immune activation and endothelial dysfunction in lung disease [63]. Taken together, these results provide evidence of the potential usefulness of MIF as a non-invasive biomarker that could be helpful in measuring the burden of disease, response to therapy, and risk of relapse in chronic respiratory disease. Preclinical and translational research has also indicated that pharmacological regulation of MIF signalling can suppress the synthesis of inflammatory cytokines, inhibit tissue remodelling and re-establish immune regulatory homeostasis in experimental pulmonary disease models, and that MIF-based interventions may be useful in respiratory medicine [17, 22]. Although several MIF inhibitors have demonstrated promising anti-inflammatory and antifibrotic effects in experimental models, clinical translation remains limited. Most compounds, including ISO-1, 4-IPP, and CPSI-1306, have primarily been evaluated in preclinical settings, and further investigation is required to determine their therapeutic feasibility and safety in pulmonary diseases.
Therapeutic targeting strategies
Therapeutic modulation of MIF signalling represents an emerging area of translational research. Several alternatives have been considered in order to inhibit the expression or the signalling of MIF, among them being small-molecule inhibitors such as ISO-1, 4-IPP, and CPSI-1306 that block MIF tautomerase activity or receptor binding. Pharmacological interference with MIF-induced inflammasome activation has been shown to restore autophagic functions and inhibit the expression of proinflammatory cytokines to restore immune regulatory responses [64]. The MIF antagonism of the oncological models causes inhibition of angiogenic and metastatic signalling, supporting therapeutic significance in inflammatory and neoplastic pulmonary pathology [65]. At the same time, monoclonal antibodies targeting CD74 or CXCR4 receptors, as well as receptor antagonists, have demonstrated activity in preclinical respiratory models. Certain natural bioactive compounds, including green tea polyphenols, have also been proposed as adjunctive modulators capable of attenuating MIF-mediated signalling through redox-sensitive mechanisms [66]. Since pulmonary immunity is compartmentalised, lung-selective delivery strategies are increasingly being explored to limit systemic immunosuppression. Inhalable formulations, aerosolised small-molecule inhibitors, and nanoparticle-based delivery platforms may increase local drug concentrations within the alveolar and airway microenvironment while reducing systemic exposure and off-target adverse effects [67, 68]. Pulmonary pharmacokinetic optimisation and tissue retention are therefore required to ensure adequate receptor occupancy and effective modulation of inflammatory signalling circuits [69, 70]. Innovative drug-delivery strategies such as nanocarriers, controlled-release systems, and inhalation-based therapeutic platforms may enable localised immune modulation while preserving systemic immune function [71, 72]. This spatially confined intervention may be particularly relevant in chronic inflammatory lung diseases where sustained local MIF amplification contributes to structural remodelling, oxidative imbalance, and steroid resistance [73, 74]. The concept of targeted pulmonary delivery of pharmacokinetically optimised molecules within a systems-level framework of immune regulation may enhance therapeutic efficacy while minimising disruption of host defence mechanisms. Table 5 summarises representative therapeutic strategies targeting MIF signalling and their current stage of translational development in pulmonary disease models.
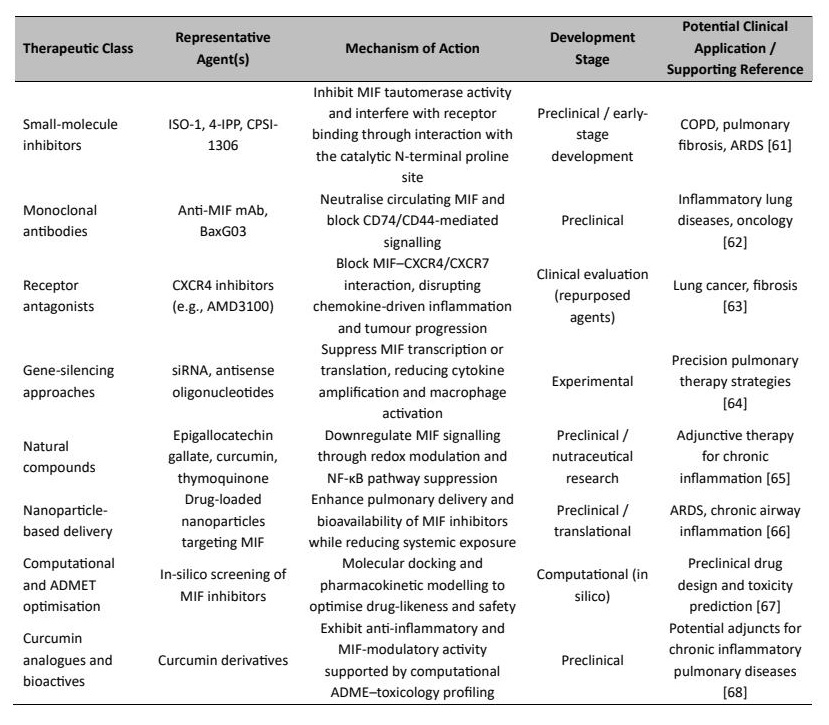
Table 5: Representative Preclinical and Emerging Therapeutic Strategies Targeting MIF Signalling in Pulmonary Diseases
Challenges and prospects
In spite of the encouraging progress achieved, MIF-targeted therapies face several challenges to clinical application. MIF and D-dopachrome tautomerase (DDT) have structural homology and partial functional redundancy, which is a significant limitation and complicates the selection of which one to inhibit. Inter-individual variability that is facilitated by MIF promoter polymorphisms further influences the therapeutic responsiveness and adverse immune modulation. Moreover, the cytokine networks relate to each other, and the expression patterns of interleukin can be heterogeneous, thus it may be challenging to predict the outcome of therapy and consequently the precision-oriented approaches are mandatory [75]. The future clinical development programs must therefore include pharmacogenomic profiling, in the future which includes the longitudinal monitoring of the biomarkers to guide the stratification of a patient and optimisation of the treatment [76]. The MIF genotype-based individualised medicine plans, determining receptor forms, dynamic immune profiles and receptor forms, can enhance the maximal therapeutic responses and reduce the systemic immune perturbation. These integrative models are consistent with MIF targeting and personalised pulmonary care and disease management at the systems level [77].
Pharmacokinetic, ADMET, and Delivery Considerations in MIF-Targeted Therapeutics
The pulmonary inflammatory disorders entail compartmentalised immunity in the airways and alveoli tissues; hence, the strategy of the pharmacokinetic optimisation and lung-target delivery is especially crucial to transfer macrophage migration inhibitory factor (MIF)-targeted therapeutic agents into clinical practice. Since MIF has many immunoregulatory activities, non-selective inhibition can disrupt physiological immune homeostasis. Molecular drug disposition needs to be coupled with immune regulation dynamics thus generating selective modulation of MIF signalling and not selective inhibition. Pharmacokinetic methods that target the lung are specifically significant in ensuring a high performance and low systemic immune suppression (Table 6).
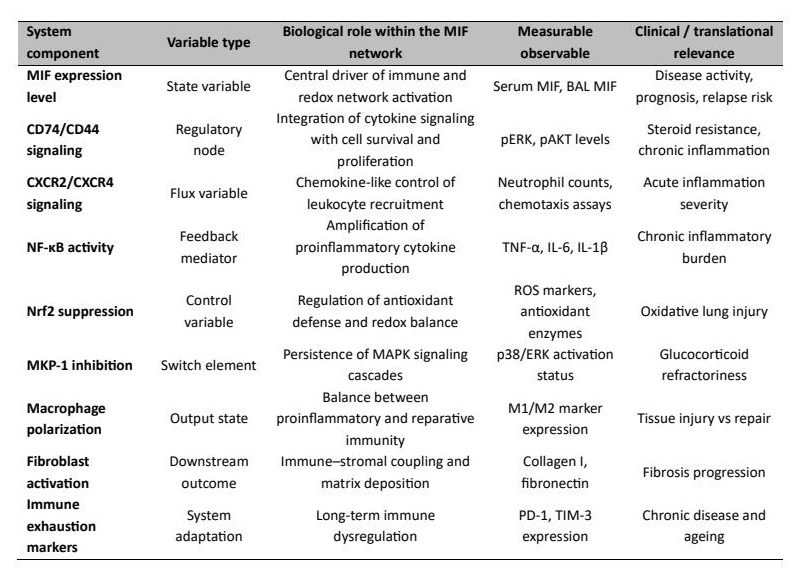
Table 6: Systems-level variables and observables in MIF signalling relevant to pulmonary disease
Absorption and Distribution
Numerous MIF inhibitors have poor oral bioavailability because of low aqueous solubility and poor membrane permeability [85-87]. Small-molecule tautomerase inhibitors like ISO-1 and 4-IPP also exhibit lower bioavailability with the use of traditional systemic route [88, 89]. This has led to the development of other delivery methods to enhance pulmonary delivery and tissue exposure of such compounds. Pulmonary drug bioavailability can be improved, local drug levels in airway and alveolar chambers can be elevated, and systemic exposure can be minimized in inflammatory diseases of the lungs, by the use of inhalable formulations and nanoparticle-based delivery systems [90-93]. Inhalation-based delivery approaches are particularly advantageous because they allow direct deposition within pulmonary tissues while limiting systemic toxicity and off-target immune suppression. Preclinical pharmacokinetic studies, therefore, emphasise maintaining effective pulmonary drug concentrations while minimising systemic exposure [94-96].
Metabolism and Biotransformation
The metabolism of several candidate MIF inhibitors involves cytochrome P450 enzymes, particularly CYP3A4 and CYP2C9, which may contribute to relatively short elimination half-lives [97-99]. Medicinal chemistry optimisation strategies have therefore been explored to improve metabolic stability and prolong drug exposure within pulmonary tissues [100, 101].
ADME–Toxicology and Safety Profiling
Safety assessment remains an essential component of cytokine-modulating therapies. Excessive suppression of MIF signalling may disrupt redox balance, mitochondrial function, and immune defence mechanisms [102, 103]. Strategies involving controlled or partial inhibition of MIF activity aim to preserve beneficial immunoregulatory responses while minimising oxidative imbalance and off-target toxicity. Nanoparticle-based delivery systems may further improve safety by concentrating drug exposure within pulmonary tissues while reducing systemic accumulation [104, 105]. Collectively, integration of pharmacokinetic optimisation, ADMET profiling, and lung-targeted delivery approaches is essential for translating MIF inhibitors into potential therapeutic strategies currently under preclinical investigation for pulmonary inflammatory and fibrotic diseases.
Future Directions
The development of the body of knowledge about the macrophage migration inhibitory factor (MIF) as a controller of the progression of pulmonary diseases justifies the necessity of integrative research strategies that combine the methods of molecular, cellular, and computational research. The heterogeneity and context dependence of MIF-associated signalling programs in the complex lung microenvironment could be overcome with multi-omics approaches, such as single-cell transcriptomics, proteomics and metabolomics [106]. Analysis of immune and stromal cell population in high-resolution can be employed to determine cell-specific MIF-regulated states which are undetectable in bulk-based analyses and multi-scale modelling and integrative mapping of cytokine interaction networks are also likely to enhance the identification of pathogenic MIF-dependent modules and new therapeutic entry points in fibrotic and inflammatory lung diseases [107]. Another significant aspect that governs MIF signalling is age-related modifications in immune homeostasis and metabolic control. MIF is often upregulated in senescence-induced immune cells, facilitating long-lasting low-grade inflammation and redox imbalance of mitochondrion which may support maladaptive signalling in the course of fibrotic progression when the body is subjected to oxidative and metabolic stress [108]. More specific MIF signalling control in this situation can be used to help re-establish metabolic balance, curb inflammatory amplification in the chronic phase and boost immune vigilance in ageing lung parenchyma. Parallel therapeutic repurposing approaches and combination therapies such as MIF inhibitors with corticosteroids, antioxidant therapies or with existing antifibrotic drugs can potentially have synergistic therapeutic effects with reduced systemic toxicity [109]. Finally, precision pulmonology approaches integrating patient-specific MIF expression profiles, promoter polymorphisms, and metabolomic signatures may provide a framework for individualised therapeutic strategies in heterogeneous pulmonary disease phenotypes.
Conclusion
Macrophage migration inhibitory factor (MIF) has become a focal regulatory mediator in pulmonary immunity with versatile and context-dependent molecular and cellular functions. MIF was initially described as a pro-inflammatory cytokine, but is currently known to have two effects: to promote host defence in acute infection and tissue damage, as well as to promote chronic inflammation, fibrotic remodelling, and tumourigenesis when control is lost. These findings highlight the role of MIF as a central regulatory hub coordinating pulmonary immune responses. MIF plays a central role in mediating the interconnectedness of immune defence response in relation to metabolic and oxidative control in the lung by coordinated effects on the activation of macrophages, T-cell differentiation, redox signalling, and epithelial integrity. The growing body of evidence supports MIF as a diagnostic and prognostic biomarker, and the levels of this protein circulating in the blood, as well as tissue-associated, are associated with disease activity and progression in chronic obstructive lung disease, idiopathic pulmonary fibrosis, and lung cancer. Simultaneous progress and development in therapeutic approaches, including small-molecule inhibitors, monoclonal antibodies, and receptor-targeted antagonists against MIF or its homolog D-dopachrome tautomerase, have increased the prospects of pharmacological intervention. Nevertheless, issues of target selectivity, potential systemic immunosuppression, and inter-individual genetic variability continue to challenge clinical translation. Future therapeutic strategies will likely rely on lung-targeted pharmacological modulation of MIF signalling combined with precision-medicine approaches to optimise efficacy while preserving protective immune functions. Integrative approaches involving omics-based profiling, systems pharmacology, and precision-guided clinical strategies will therefore be critical for advancing MIF-directed pulmonary medicine. It will be necessary to clarify the contextual determinants by which MIF activity is either protective or transitions to pathogenic signalling to selectively utilise its beneficial functions while limiting pathological effects. A deeper understanding of the regulatory networks governing MIF-driven immune responses may ultimately enable mechanism-based and personalised therapeutic strategies for complex pulmonary diseases.
Acknowledgements
The authors would like to thank Azerbaijan Medical University for providing institutional and academic support during the preparation of this manuscript. The authors also acknowledge the support of the Departments of Cytology, Embryology and Histology, Public Health and Healthcare Organization, Pharmaceutical Toxicology and Chemistry, and Human Anatomy and Medical Terminology of Azerbaijan Medical University, Baku.
Author Contributions
Aliyarbayova A.A. conceptualized the study, designed the structure of the review, supervised manuscript development, contributed to scientific interpretation, and served as corresponding author. Pashayeva S.A. contributed to literature acquisition, critical analysis of molecular and pharmacological sections, and drafting of relevant manuscript sections. Qaniyeva G.M. contributed to the anatomical and immunological contextualization of pulmonary mechanisms and assisted in drafting and revising the manuscript. Qarayeva S.D. participated in literature review, data interpretation related to pulmonary pathology, and manuscript editing. Hajiyeva Y.A. assisted in manuscript preparation, reference organisation, formatting, and figure/table coordination. Mekhtiyeva A.F. contributed to literature compilation, technical editing, and preparation of the final manuscript version.
Funding Sources
The authors declare that no specific external funding was received for this study.
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors have no conflicts of interest to declare.
References
- Aliyarbayova A, Sultanova T, Yaqubova S, Najafova T, Sadiqova G, Salimov A a. Macrophage Migration Inhibitory Factor: Its Multifaceted Role in Inflammation and Immune Regulation Across Organ Systems. Cell Physiol Biochem. 2025 Sep 3;59(5):569-88. https://doi.org/10.33594/000000809
- Kang I, Bucala R. The immunobiology of MIF: function, genetics and prospects for precision medicine. Nat Rev Rheumatol. 2019 Jul;15(7):427-37.https://doi.org/10.1038/s41584-019-0238-2
- Harris J, VanPatten S, Deen NS, Al-Abed Y, Morand EF. Rediscovering MIF: New Tricks for an Old Cytokine. Trends in Immunology. 2019 May;40(5):447-62.https://doi.org/10.1016/j.it.2019.03.002
- Sinitski D, Kontos C, Krammer C, Asare Y, Kapurniotu A, Bernhagen J. Macrophage Migration Inhibitory Factor (MIF)-Based Therapeutic Concepts in Atherosclerosis and Inflammation. Thromb Haemost. 2019 Apr;119(04):553-66.https://doi.org/10.1055/s-0039-1677803
- Petralia MC, Battaglia G, Bruno V, Pennisi M, Mangano K, Lombardo SD, et al. The Role of Macrophage Migration Inhibitory Factor in Alzheimer′s Disease: Conventionally Pathogenetic or Unconventionally Protective? Molecules. 2020 Jan 10;25(2):291.https://doi.org/10.3390/molecules25020291
- Leyton-Jaimes MF, Kahn J, Israelson A. Macrophage migration inhibitory factor: A multifaceted cytokine implicated in multiple neurological diseases. Experimental Neurology. 2018 Mar;301:83-91.https://doi.org/10.1016/j.expneurol.2017.06.021
- Bucala R, Bernhagen J, editors. MIF Family Cytokines in Innate Immunity and Homeostasis [Internet]. Cham: Springer International Publishing; 2017 [cited 2026 Feb 12]. Available from: http://link.springer.com/10.1007/978-3-319-52354-5https://doi.org/10.1007/978-3-319-52354-5
- Leng L, Siu E, Bucala R. Genotyping Two Promoter Polymorphisms in the MIF Gene: A −794 CATT5-8 Microsatellite Repeat and a −173 G/C SNP. In: Harris J, Morand EF, editors. Macrophage Migration Inhibitory Factor [Internet]. New York, NY: Springer US; 2020 [cited 2026 Feb 12]. p. 67-(Methods in Molecular Biology; vol. 2080). Available from: http://link.springer.com/10.1007/978-1-4939-9936-1_7https://doi.org/10.1007/978-1-4939-9936-1_7
- Illescas O, Gomez-Verjan JC, García-Velázquez L, Govezensky T, Rodriguez-Sosa M. Macrophage Migration Inhibitory Factor -173 G/C Polymorphism: A Global Meta-Analysis across the Disease Spectrum. Front Genet. 2018 Mar 1;9:55.https://doi.org/10.3389/fgene.2018.00055
- Hizawa N, Yamaguchi E, Takahashi D, Nishihira J, Nishimura M. Functional Polymorphisms in the Promoter Region of Macrophage Migration Inhibitory Factor and Atopy. Am J Respir Crit Care Med. 2004 May 1;169(9):1014-8.https://doi.org/10.1164/rccm.200307-933OC
- Florez-Sampedro L, Soto-Gamez A, Poelarends GJ, Melgert BN. The role of MIF in chronic lung diseases: looking beyond inflammation. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2020 Jun 1;318(6):L1183-97.https://doi.org/10.1152/ajplung.00521.2019
- Cebulla CM, Stevenson W, Van Law H, Heisler-Taylor T, Hamadmad S, Shah MH, et al. MIF promoter polymorphisms are associated with epiretinal membrane but not retinal detachment with PVR in an American population. Experimental Eye Research. 2019 Aug;185:107667.https://doi.org/10.1016/j.exer.2019.05.007
- Ma M, Tao L, Liu A, Liang Z, Yang J, Peng Y, et al. Macrophage migration inhibitory factor-794 CATT microsatellite polymorphism and risk of tuberculosis: a meta-analysis. Bioscience Reports. 2018 Aug 31;38(4):BSR20171626.https://doi.org/10.1042/BSR20171626
- Cakan N, Yılmaz R, Karaaslan E, Ateş Ö. Association of Macrophage Migration Inhibitory Factor Gene -173 G/C Polymorphism (rs755622) with Familial Mediterranean Fever in Children. J Pediatr Genet. 2022 Jun;11(02):091-8.https://doi.org/10.1055/s-0040-1719053
- Kok T, Wasiel AA, Cool RH, Melgert BN, Poelarends GJ, Dekker FJ. Small-molecule inhibitors of macrophage migration inhibitory factor (MIF) are an emerging class of therapeutics for immune disorders. Drug Discovery Today. 2018 Nov;23(11):1910-8.https://doi.org/10.1016/j.drudis.2018.06.017
- Trivedi-Parmar V, Jorgensen WL. Advances and Insights for Small Molecule Inhibition of Macrophage Migration Inhibitory Factor. J Med Chem. 2018 Sep 27;61(18):8104-19.https://doi.org/10.1021/acs.jmedchem.8b00589
- Guo S, Zhao Y, Yuan Y, Liao Y, Jiang X, Wang L, et al. Progress in the development of macrophage migration inhibitory factor small-molecule inhibitors. European Journal of Medicinal Chemistry. 2025 Mar;286:117280.https://doi.org/10.1016/j.ejmech.2025.117280
- Fan Z, Wang H, Pan J, Yu S, Xia W. Potential role of macrophage migration inhibitory factor in the pathogenesis of Marek's disease. Journal of Veterinary Research. 2020 Jan 31;64(1):33-8.https://doi.org/10.2478/jvetres-2020-0009
- Husebø GR, Bakke PS, Grønseth R, Hardie JA, Ueland T, Aukrust P, et al. Macrophage migration inhibitory factor: a role in COPD. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2016 Jul 1;311(1):L1-7.https://doi.org/10.1152/ajplung.00461.2015
- Bargagli E, Olivieri C, Nikiforakis N, Cintorino M, Magi B, Perari MG, et al. Analysis of macrophage migration inhibitory factor (MIF) in patients with idiopathic pulmonary fibrosis. Respiratory Physiology & Neurobiology. 2009 Jul;167(3):261-7.https://doi.org/10.1016/j.resp.2009.05.004
- Jalce G, Guignabert C. Multiple roles of macrophage migration inhibitory factor in pulmonary hypertension. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2020 Jan 1;318(1):L1-9.https://doi.org/10.1152/ajplung.00234.2019
- Lan H, Wang N, Chen Y, Wang X, Gong Y, Qi X, et al. Macrophage migration inhibitory factor (MIF) promotes rat airway muscle cell proliferation and migration mediated by ERK1/2 and FAK signalling. Cell Biology International. 2018 Jan;42(1):75-83.https://doi.org/10.1002/cbin.10863
- Lan H, Luo L, Chen Y, Wang M, Yu Z, Gong Y. MIF signalling blocking alleviates airway inflammation and airway epithelial barrier disruption in a HDM-induced asthma model. Cellular Immunology. 2020 Jan;347:103965.https://doi.org/10.1016/j.cellimm.2019.103965
- Ahmed M, Miller E. Macrophage migration inhibitory factor (MIF) in the development and progression of pulmonary arterial hypertension. gcsp [Internet]. 2018 Jul 23 [cited 2026 Feb 12];2018(2). Available from: https://globalcardiologyscienceandpractice.com/index.php/gcsp/article/view/317https://doi.org/10.21542/gcsp.2018.14
- Chen X, Tang J, Shuai W, Meng J, Feng J, Han Z. Macrophage polarization and its role in the pathogenesis of acute lung injury/acute respiratory distress syndrome. Inflamm Res. 2020 Sep;69(9):883-95.https://doi.org/10.1007/s00011-020-01378-2
- Aegerter H, Lambrecht BN, Jakubzick CV. Biology of lung macrophages in health and disease. Immunity. 2022 Sep;55(9):1564-80.https://doi.org/10.1016/j.immuni.2022.08.010
- Bueno M, Lai YC, Romero Y, Brands J, St. Croix CM, Kamga C, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. 2015 Feb 2;125(2):521-38.https://doi.org/10.1172/JCI74942
- Mehta M, Dhanjal DS, Paudel KR, Singh B, Gupta G, Rajeshkumar S, et al. Cellular signalling pathways mediating the pathogenesis of chronic inflammatory respiratory diseases: an update. Inflammopharmacol. 2020 Aug;28(4):795-817.https://doi.org/10.1007/s10787-020-00698-3
- Checa J, Aran JM. Airway Redox Homeostasis and Inflammation Gone Awry: From Molecular Pathogenesis to Emerging Therapeutics in Respiratory Pathology. IJMS. 2020 Dec 7;21(23):9317.https://doi.org/10.3390/ijms21239317
- Mumby S, Chung KF, Adcock IM. Transcriptional Effects of Ozone and Impact on Airway Inflammation. Front Immunol. 2019 Jul 10;10:1610.https://doi.org/10.3389/fimmu.2019.01610
- Wiegman CH, Li F, Ryffel B, Togbe D, Chung KF. Oxidative Stress in Ozone-Induced Chronic Lung Inflammation and Emphysema: A Facet of Chronic Obstructive Pulmonary Disease. Front Immunol. 2020 Sep 2;11:1957.https://doi.org/10.3389/fimmu.2020.01957
- Ma WT, Gao F, Gu K, Chen DK. The Role of Monocytes and Macrophages in Autoimmune Diseases: A Comprehensive Review. Front Immunol. 2019 May 24;10:1140.https://doi.org/10.3389/fimmu.2019.01140
- Das R, Koo MS, Kim BH, Jacob ST, Subbian S, Yao J, et al. Macrophage migration inhibitory factor (MIF) is a critical mediator of the innate immune response to Mycobacterium tuberculosis. Proc Natl Acad Sci USA [Internet]. 2013 Aug 6 [cited 2026 Feb 12];110(32). Available from: https://pnas.org/doi/full/10.1073/pnas.1301128110https://doi.org/10.1073/pnas.1301128110
- Hoffmann RF, Zarrintan S, Brandenburg SM, Kol A, de Bruin HG, Jafari S, Dijk F, Kalicharan D, Kelders M, Gosker HR, Ten Hacken NH, van der Want JJ, van Oosterhout AJ, Heijink IH. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res. 2013 Oct 2;14(1):97.https://doi.org/10.1186/1465-9921-14-97
- Prasse A, Binder H, Schupp JC, Kayser G, Bargagli E, Jaeger B, Hess M, Rittinghausen S, Vuga L, Lynn H, Violette S, Jung B, Quast K, Vanaudenaerde B, Xu Y, Hohlfeld JM, Krug N, Herazo-Maya JD, Rottoli P, Wuyts WA, Kaminski N. BAL Cell Gene Expression Is Indicative of Outcome and Airway Basal Cell Involvement in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2019 Mar 1;199(5):622-630.https://doi.org/10.1164/rccm.201712-2551OC
- Li Q, Zheng H, Chen B. Identification of macrophage-related genes in sepsis-induced ARDS using bioinformatics and machine learning. Sci Rep. 2023;13:9876.https://doi.org/10.1038/s41598-023-37162-5
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Pontén F. Proteomics. Tissue-based map of the human proteome. Science. 2015 Jan 23;347(6220):1260419.https://doi.org/10.1126/science.1260419
- Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, Jensen LJ, von Mering C. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021 Jan 8;49(D1):D605-D6doi: 10.1093/nar/gkaa1074. Erratum in: Nucleic Acids Res. 2021 Oct 11;49(18):10800.https://doi.org/10.1093/nar/gkab835
- Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, Suveges D, Vrousgou O, Whetzel PL, Amode R, Guillen JA, Riat HS, Trevanion SJ, Hall P, Junkins H, Flicek P, Burdett T, Hindorff LA, Cunningham F, Parkinson H. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019 Jan 8;47(D1):D1005-D1012.https://doi.org/10.1093/nar/gky1120
- Li J, Ma W, Tang Z, Li Y, Zheng R, Xie Y, et al. Macrophage‑driven pathogenesis in acute lung injury/acute respiratory disease syndrome: Harnessing natural products for therapeutic interventions (Review). Mol Med Rep. 2024 Nov 4;31(1):16.https://doi.org/10.3892/mmr.2024.13381
- De Souza GF, Muraro SP, Santos LD, Monteiro APT, Da Silva AG, De Souza APD, et al. Macrophage migration inhibitory factor (MIF) controls cytokine release during respiratory syncytial virus infection in macrophages. Inflamm Res. 2019 Jun;68(6):481-91.https://doi.org/10.1007/s00011-019-01233-z
- Nieto-Fontarigo JJ, González-Barcala FJ, San José E, Arias P, Nogueira M, Salgado FJ. CD26 and Asthma: a Comprehensive Review. Clinic Rev Allerg Immunol. 2019 Apr 15;56(2):139-60.https://doi.org/10.1007/s12016-016-8578-z
- Bianco A, Perrotta F, Barra G, Malapelle U, Rocco D, De Palma R. Prognostic Factors and Biomarkers of Responses to Immune Checkpoint Inhibitors in Lung Cancer. IJMS. 2019 Oct 5;20(19):4931.https://doi.org/10.3390/ijms20194931
- Naccache JM, Gibiot Q, Monnet I, Antoine M, Wislez M, Chouaid C, et al. Lung cancer and interstitial lung disease: a literature review. J Thorac Dis. 2018 Jun;10(6):3829-44.https://doi.org/10.21037/jtd.2018.05.75
- Yang P, Luo Q, Wang X, Fang Q, Fu Z, Li J, et al. Comprehensive Analysis of Fibroblast Activation Protein Expression in Interstitial Lung Diseases. American Journal of Respiratory and Critical Care Medicine. 2023 Jan 15;207(2):160-72.https://doi.org/10.1164/rccm.202110-2414OC
- Kishore A, Petrek M. Roles of Macrophage Polarization and Macrophage-Derived miRNAs in Pulmonary Fibrosis. Front Immunol. 2021 Aug 13;12:678457.https://doi.org/10.3389/fimmu.2021.678457
- Jiang M, Bu W, Wang X, Ruan J, Shi W, Yu S, et al. Pulmonary fibrosis: from mechanisms to therapies. J Transl Med. 2025 May 8;23(1):515.https://doi.org/10.1186/s12967-025-06514-2
- Heinrichs D, Knauel M, Offermanns C, Berres ML, Nellen A, Leng L, et al. Macrophage migration inhibitory factor (MIF) exerts antifibrotic effects in experimental liver fibrosis via CD74. Proc Natl Acad Sci USA. 2011 Oct 18;108(42):17444-9.https://doi.org/10.1073/pnas.1107023108
- Datta A, Scotton CJ, Chambers RC. Novel therapeutic approaches for pulmonary fibrosis. British J Pharmacology. 2011 May;163(1):141-72.https://doi.org/10.1111/j.1476-5381.2011.01247.x
- Günther S, Bordenave J, Hua-Huy T, Nicco C, Cumont A, Thuillet R, et al. Macrophage Migration Inhibitory Factor (MIF) Inhibition in a Murine Model of Bleomycin-Induced Pulmonary Fibrosis. IJMS. 2018 Dec 18;19(12):4105.https://doi.org/10.3390/ijms19124105
- Günther S, Fagone P, Jalce G, Atanasov AG, Guignabert C, Nicoletti F. Role of MIF and D-DT in immune-inflammatory, autoimmune, and chronic respiratory diseases: from pathogenic factors to therapeutic targets. Drug Discovery Today. 2019 Feb;24(2):428-39.https://doi.org/10.1016/j.drudis.2018.11.003
- Bernhagen J. A new cytokine target for chronic obstructive pulmonary disease? eBioMedicine. 2021 Jul;69:103479.https://doi.org/10.1016/j.ebiom.2021.103479
- Plichta J, Kuna P, Panek M. Biologic drugs in the treatment of chronic inflammatory pulmonary diseases: recent developments and future perspectives. Front Immunol. 2023 Jun 2;14:1207641.https://doi.org/10.3389/fimmu.2023.1207641
- Arora P, Nainwal LM, Athari SS. Editorial: Molecular pharmacological approaches against lung diseases: targeted drug discovery. Front Pharmacol. 2024 Jul 9;15:1419138.https://doi.org/10.3389/fphar.2024.1419138
- Brewer KD, Santo NV, Samanta A, Nag R, Trotsyuk AA, Rajadas J. Advances in Therapeutics for Chronic Lung Diseases: From Standard Therapies to Emerging Breakthroughs. JCM. 2025 Apr 30;14(9):3118.https://doi.org/10.3390/jcm14093118
- Kumar V, Mundra V, Peng Y, Wang Y, Tan C, Mahato RI. Pharmacokinetics and biodistribution of polymeric micelles containing miRNA and small-molecule drug in orthotopic pancreatic tumor-bearing mice. Theranostics. 2018;8(15):4033-49.https://doi.org/10.7150/thno.24945
- Charan M, Das S, Mishra S, Chatterjee N, Varikuti S, Kaul K, et al. Macrophage migration inhibitory factor inhibition as a novel therapeutic approach against triple-negative breast cancer. Cell Death Dis. 2020 Sep 17;11(9):774.https://doi.org/10.1038/s41419-020-02992-y
- Grandoni S. A PHYSIOLOGICALLY BASED PHARMACOKINETIC MODELLING FRAMEWORK TO SUPPORT INHALED DRUG DEVELOPMENT FROM THE EARLY TO THE LATE STAGES. 2020 [cited 2026 Feb 12]; Available from: https://iris.unipv.it/handle/11571/1315927
- Gurunathan S, Kang MH, Kim JH. A Comprehensive Review on Factors Influences Biogenesis, Functions, Therapeutic and Clinical Implications of Exosomes. IJN. 2021 Feb;Volume 16:1281-312.https://doi.org/10.2147/IJN.S291956
- Mitra S, Paul S, Roy S, Sutradhar H, Bin Emran T, Nainu F, et al. Exploring the Immune-Boosting Functions of Vitamins and Minerals as Nutritional Food Bioactive Compounds: A Comprehensive Review. Molecules. 2022 Jan 16;27(2):555.https://doi.org/10.3390/molecules27020555
- Hannan MdA, Rahman MdA, Sohag AAM, Uddin MdJ, Dash R, Sikder MH, et al. Black Cumin (Nigella sativa L.): A Comprehensive Review on Phytochemistry, Health Benefits, Molecular Pharmacology, and Safety. Nutrients. 2021 May 24;13(6):1784.https://doi.org/10.3390/nu13061784
- Soulaidopoulos S. Pulmonary manifestations of chronic liver disease: a comprehensive review. aog [Internet]. 2020 [cited 2026 Feb 12]; Available from: http://www.annalsgastro.gr/files/journals/1/earlyview/2020/ev-04-2020-07-AG4818-0474.pdf
- Mohan A, Agarwal S, Clauss M, Britt NS, Dhillon NK. Extracellular vesicles: novel communicators in lung diseases. Respir Res. 2020 Dec;21(1):175.https://doi.org/10.1186/s12931-020-01423-y
- Liu S, Zhang J. A comprehensive analysis of the relationship between inflammasomes and autophagy in human tumors: Recent developments. Journal of Cell Communication and Signalling. 2025 Sep;19(3):e70035.https://doi.org/10.1002/ccs3.70035
- Soumoy L, Kindt N, Ghanem G, Saussez S, Journe F. Role of Macrophage Migration Inhibitory Factor (MIF) in Melanoma. Cancers. 2019 Apr 12;11(4):529.https://doi.org/10.3390/cancers11040529
- Tallei TE, Fatimawali, Niode NJ, Idroes R, Zidan BMRM, Mitra S, et al. A Comprehensive Review of the Potential Use of Green Tea Polyphenols in the Management of COVID-19. De La Puerta R, editor. Evidence-Based Complementary and Alternative Medicine. 2021 Dec 3;2021:1-13.https://doi.org/10.1155/2021/7170736
- Ouertatani-Sakouhi H, El-Turk F, Fauvet B, Cho MK, Pinar Karpinar D, Le Roy D, et al. Identification and Characterization of Novel Classes of Macrophage Migration Inhibitory Factor (MIF) Inhibitors with Distinct Mechanisms of Action. Journal of Biological Chemistry. 2010 Aug;285(34):26581-98.https://doi.org/10.1074/jbc.M110.113951
- Mizue Y, Ghani S, Leng L, McDonald C, Kong P, Baugh J, et al. Role for macrophage migration inhibitory factor in asthma. Proc Natl Acad Sci USA. 2005 Oct 4;102(40):14410-5.https://doi.org/10.1073/pnas.0507189102
- Khezrian A, Shojaeian A, Khaghani Boroujeni A, Amini R. Therapeutic Opportunities in Breast Cancer by Targeting Macrophage Migration Inhibitory Factor as a Pleiotropic Cytokine. Breast Cancer (Auckl). 2024 Jan;18:11782234241276310.https://doi.org/10.1177/11782234241276310
- Jalaie M, Arimoto R, Gifford E, Schefzick S, Waller CL. Prediction of Drug-Like Molecular Properties. In: Bajorath J, editor. Chemoinformatics [Internet]. Totowa, NJ: Humana Press; 2004 [cited 2026 Feb 12]. p. 449-5(Walker JM, editor. Methods in Molecular Biology; vol. 275). Available from: http://link.springer.com/10.1385/1-59259-802-1:449https://doi.org/10.1385/1-59259-802-1:449
- Merecz-Sadowska A, Sitarek P, Śliwiński T, Zajdel R. Anti-Inflammatory Activity of Extracts and Pure Compounds Derived from Plants via Modulation of Signalling Pathways, Especially PI3K/AKT in Macrophages. IJMS. 2020 Dec 16;21(24):9605.https://doi.org/10.3390/ijms21249605
- Doroudian M, O'Neill A, O'Reilly C, Tynan A, Mawhinney L, McElroy A, et al. Aerosolized Drug-Loaded Nanoparticles Targeting Migration Inhibitory Factors Inhibit Pseudomonas Aeruginosa -Induced Inflammation and Biofilm Formation. Nanomedicine (Lond). 2020 Dec;15(30):2933-53.https://doi.org/10.2217/nnm-2020-0344
- Nagarajan P, Louis LRP, Rangarajalu K. Targeting Macrophage Migration Inhibitory Factor (Mif): Small-Molecule Inhibitors And Therapeutic Strategies For Immune Disorders And Cancer. Gomal Journal of Medical Sciences [Internet]. 2024 [cited 2026 Feb 12];22(3).
- Chabib L, Awaluddin R, Ikawati Z, Martien R, Ismail H. Molecular Docking, Pharmacophore Modelling, And Adme-Toxicity Prediction Of Curcumin Analog Compounds As Inflammatory Inhibitor On Rheumatoid Arthritis. Int J Pharm Pharm Sci. 2017 Jul 22;9(9):16.https://doi.org/10.22159/ijpps.2017v9i9.20450
- Al-Qahtani AA, Alhamlan FS, Al-Qahtani AA. Pro-Inflammatory and Anti-Inflammatory Interleukins in Infectious Diseases: A Comprehensive Review. TropicalMed. 2024 Jan 4;9(1):13.https://doi.org/10.3390/tropicalmed9010013
- Døllner H, Vatten L, Halgunset J, Rahimipoor S, Austgulen R. Histologic chorioamnionitis and umbilical serum levels of pro‐inflammatory cytokines and cytokine inhibitors. BJOG. 2002 May;109(5):534-9.https://doi.org/10.1111/j.1471-0528.2002.01028.x
- Baugh JA, Bucala R. Macrophage migration inhibitory factor. Critical care medicine. 2002;30(1):S27-35.https://doi.org/10.1097/00003246-200201001-00004
- Orita M, Yamamoto S, Katayama N, Fujita S. Macrophage Migration Inhibitory Factor and the Discovery of Tautomerase Inhibitors. CPD. 2002 Jun 1;8(14):1297-317.https://doi.org/10.2174/1381612023394674
- Xu L, Li Y, Sun H, Zhen X, Qiao C, Tian S, et al. Current developments of macrophage migration inhibitory factor (MIF) inhibitors. Drug Discovery Today. 2013 Jun;18(11-12):592-600.https://doi.org/10.1016/j.drudis.2012.12.013
- Höllriegl W, Bauer A, Baumgartner B, Dietrich B, Douillard P, Kerschbaumer RJ, et al. Pharmacokinetics, disease-modifying activity, and safety of an experimental therapeutic targeting an immunological isoform of macrophage migration inhibitory factor, in rat glomerulonephritis. European Journal of Pharmacology. 2018 Feb;820:206-16.https://doi.org/10.1016/j.ejphar.2017.12.040
- Williams RO, Paleolog E, Feldmann M. Cytokine inhibitors in rheumatoid arthritis and other autoimmune diseases. Current Opinion in Pharmacology. 2007 Aug;7(4):412-7.https://doi.org/10.1016/j.coph.2007.06.001
- Wang J, Urban L. The impact of early ADME profiling on drug discovery and development strategy. DDW Drug Discovery World. 2004;5(4):73-86.
- Balachandran S, Gadekar PK, Parkale S, Yadav VN, Kamath D, Ramaswamy S, et al. Synthesis and biological activity of novel MIF antagonists. Bioorganic & Medicinal Chemistry Letters. 2011 Mar;21(5):1508-11.https://doi.org/10.1016/j.bmcl.2010.12.127
- Cournia Z, Leng L, Gandavadi S, Du X, Bucala R, Jorgensen WL. Discovery of Human Macrophage Migration Inhibitory Factor (MIF)-CD74 Antagonists via Virtual Screening. J Med Chem. 2009 Jan 22;52(2):416-24.https://doi.org/10.1021/jm801100v
- Kallen A. Computational Pharmacokinetics [Internet]. 0 ed. Chapman and Hall/CRC; 2007 [cited 2026 Feb 12]. Available from: https://www.taylorfrancis.com/books/9781420060669
- Wang X, Song K, Li L, Chen L. Structure-Based Drug Design Strategies and Challenges. CTMC. 2018 Sep 18;18(12):998-1006.https://doi.org/10.2174/1568026618666180813152921
- Morris GM, Lim-Wilby M. Molecular Docking. In: Kukol A, editor. Molecular Modeling of Proteins [Internet]. Totowa, NJ: Humana Press; 2008 [cited 2026 Feb 12]. p. 365-(Walker JM, editor. Methods in Molecular Biology; vol. 443). Available from: http://link.springer.com/10.1007/978-1-59745-177-2_19https://doi.org/10.1007/978-1-59745-177-2_19
- Van Montfort RLM, Workman P. Structure-based drug design: aiming for a perfect fit. Van Montfort RLM, Workman P, editors. Essays in Biochemistry. 2017 Nov 8;61(5):431-7.https://doi.org/10.1042/EBC20170052
- Bloom J, Sun S, Al-Abed Y. MIF, a controversial cytokine: a review of structural features, challenges, and opportunities for drug development. Expert Opinion on Therapeutic Targets. 2016 Dec;20(12):1463-75.https://doi.org/10.1080/14728222.2016.1251582
- Swanson K, Walther P, Leitz J, Mukherjee S, Wu JC, Shivnaraine RV, et al. ADMET-AI: a machine learning ADMET platform for evaluation of large-scale chemical libraries. Valencia A, editor. Bioinformatics. 2024 Jul 1;40(7):btae416.https://doi.org/10.1093/bioinformatics/btae416
- Jayatunga MKP, Xie W, Ruder L, Schulze U, Meier C. AI in small-molecule drug discovery: a coming wave? Nat Rev Drug Discov. 2022 Mar;21(3):175-6.https://doi.org/10.1038/d41573-022-00025-1
- Wang W, Zhou H. Pharmacological considerations for predicting PK/PD at the site of action for therapeutic proteins. Drug Discovery Today: Technologies. 2016 Sep;21-22:35-9.https://doi.org/10.1016/j.ddtec.2016.09.006
- Kumar V, Sharma N, Maitra SS. In vitro and in vivo toxicity assessment of nanoparticles. Int Nano Lett. 2017 Dec;7(4):243-56.https://doi.org/10.1007/s40089-017-0221-3
- Larbi A, Kempf J, Pawelec G. Oxidative stress modulation and T cell activation. Experimental Gerontology. 2007 Sep;42(9):852-8.https://doi.org/10.1016/j.exger.2007.05.004
- Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C, et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Research. 2021 Jul 2;49(W1):W5-14.https://doi.org/10.1093/nar/gkab255
- Sączewski J, Popenda Ł, Fedorowicz J. In Silico SwissADME Analysis of Antibacterial NHC-Silver Acetates and Halides Complexes. Applied Sciences. 2024 Oct 2;14(19):8865.https://doi.org/10.3390/app14198865
- Danhof M. Systems pharmacology - Towards the modeling of network interactions. European Journal of Pharmaceutical Sciences. 2016 Oct;94:4-14.https://doi.org/10.1016/j.ejps.2016.04.027
- Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008 Nov;4(11):682-90.https://doi.org/10.1038/nchembio.118
- Ursu O, Rayan A, Goldblum A, Oprea TI. Understanding drug‐likeness. WIREs Comput Mol Sci. 2011 Sep;1(5):760-81.https://doi.org/10.1002/wcms.52
- Tian S, Wang J, Li Y, Li D, Xu L, Hou T. The application of in silico drug-likeness predictions in pharmaceutical research. Advanced Drug Delivery Reviews. 2015 Jun;86:2-10.https://doi.org/10.1016/j.addr.2015.01.009
- Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin II, Cronin M, et al. QSAR Modeling: Where Have You Been? Where Are You Going To? J Med Chem. 2014 Jun 26;57(12):4977-5010.https://doi.org/10.1021/jm4004285
- Skeens E, Gadzuk-Shea M, Shah D, Bhandari V, Schweppe DK, Berlow RB, et al. Redox-dependent structure and dynamics of macrophage migration inhibitory factor reveal sites of latent allostery. Structure. 2022 Jun;30(6):840-850.e6.https://doi.org/10.1016/j.str.2022.03.007
- Tripathi SC, Fahrmann JF, Vykoukal JV, Dennison JB, Hanash SM. Targeting metabolic vulnerabilities of cancer: Small molecule inhibitors in clinic. Cancer Reports. 2019 Feb;2(1):e1131.https://doi.org/10.1002/cnr2.1131
- Wang Y, Xing J, Xu Y, Zhou N, Peng J, Xiong Z, et al. In silico ADME/T modelling for rational drug design. Quart Rev Biophys. 2015 Nov;48(4):488-515.https://doi.org/10.1017/S0033583515000190
- Ancuceanu R, Lascu BE, Drăgănescu D, Dinu M. In Silico ADME Methods Used in the Evaluation of Natural Products. Pharmaceutics. 2025 Jul 31;17(8):1002.https://doi.org/10.3390/pharmaceutics17081002
- Selvarajah B, Platé M, Chambers RC. Pulmonary fibrosis: Emerging diagnostic and therapeutic strategies. Molecular Aspects of Medicine. 2023 Dec;94:101227.https://doi.org/10.1016/j.mam.2023.101227
- Schuliga M, Pechkovsky DV, Read J, Waters DW, Blokland KEC, Reid AT, et al. Mitochondrial dysfunction contributes to the senescent phenotype of IPF lung fibroblasts. J Cellular Molecular Medi. 2018 Dec;22(12):5847-61.https://doi.org/10.1111/jcmm.13855
- Rajasegaran T, How CW, Saud A, Ali A, Lim JCW. Targeting Inflammation in Non-Small Cell Lung Cancer through Drug Repurposing. Pharmaceuticals. 2023 Mar 16;16(3):451.https://doi.org/10.3390/ph16030451
- Raavi, Koehler AN, Vegas AJ. At The Interface: Small-Molecule Inhibitors of Soluble Cytokines. Chem Rev. 2025 May 14;125(9):4528-68.https://doi.org/10.1021/acs.chemrev.4c00469