Original Article – DOI: 10.33594/000000872
CPB (60): 291 - 335
Accepted: 23.06.2026 - Published: 29.06.2026
Epistemology of the Origin of Cancer IV: Predisposing Conditions for Metastases
bTheodor-Billroth-Academy® with its INCORE, International Consortium of Research Excellence, Munich, Germany, Sacramento, CA, USA
cCancer Metastases Research Fund, Sacramento, California, United States of America,
dDepartment of Surgery, Medical University Lausitz – Carl-Thiem, Cottbus, Germany
Keywords
Abstract
Causal therapy has achieved success in the treatment of epithelial tumors, which account for more than 80% of all cancers. Although frequently claimed as breakthroughs, cancer therapy has achieved limited increases in survival of only weeks to several months, and cancer incidence continues to increase and metastasis rates, which are primarily responsible for cancer mortality, remain constant. This broadly reflect an incomplete understanding of the genesis and progression of cancers and metastases. Over a 25-year timeframe, the fundamentals of the origin of carcinogenesis and metastasis, titled Epistemology of the Origin of Cancer were addressed in Part I, 2014–2022, how carcinogenesis develops; Part II, 2023, which is the first cancer cell; Part III, 2025, how metastasis arise with the formation of local pre-metastatic niches (PMN-1, PMN-2, PMN-3) and traveling cancer satellites. Herein, Part IV discusses the conditions required for metastasis: the development of distant metastatic niches (MN-1, MN-2, MN-3) after traveling, extravasation and nidation at the endothelium of the capillary bed. First, MN-1 develops by transendothelial migration of cancer satellites to the subendothelial matrix, followed by de-coating into constituent parts, induction of cell plasticity, recruitment of immuno-competent cells, and dormancy. MN-1 then transforms into MN-2 triggered by ongoing chronic inflammation, and remodeling (lysyl oxidase): pro-tumor-associated cells, extravesicular vesicles, and CXCL2 induce immune suppression, granulin, periostin, fibronectin, CXCL12, and K19 followed by migration of non-dormant cancer associated fibroblasts (CAFs) and cancer cells. Finally, MN-3 develops where high cell plasticity leads to more aggressive active elongated cancer cells and newly developed metastasis-associated fibroblasts (MAFs), which re-trigger dormancy. Anticancer therapy, surgery, trauma, and ongoing inflammation exacerbate immune suppression, which induce awakening from dormancy. Gelsolin and atypical EMT induce conversion into spherical, inactive cancer cells (enabling immune evasion), followed by another spell of dormancy. When overwhelmed, this can lead to the essential predisposing conditions for metastases by formation of metastatic cancer satellites: (1) metastatic cancer cells and (2) MAFs; (3) metastatic cancer cells surrounded by MAFs; (4) CXCL12 and K19 coated metastatic cancer cells; (5) platelets surrounding metastatic cancer cells and (6) MAFs; (7) NETs shielded metastatic cancer cells and (8) MAFs. The sequential distant metastatic niche model can serve as a coherent framework to better understand metastatic progression: metastatic cancer satellites, each with its distinct biology, are shielded from the immune system, and consequently migrate to distant sites, which fulfills the predisposing conditions for metastases. Analogously it provides biological meaning to long observed phenomena so far not satisfactorily explained: metastasis occurs concurrently with carcinogenesis; vast molecular and clinical heterogeneity; most disseminated cancer cells remain dormant for years; surgery or anticancer therapy can paradoxically trigger relapse; oncology therapies, despite direct anti-cancer cellular effects, have so far failed to substantially alter the natural history of most epithelial cancers.
Graphical Abstract – Highlights
Fig. 1. Metastatic niche 1 (MN-1) develops from pre-metastatic niches (PMNs): Cancer satellites extravasate after traveling and nidate at the capillary bed; after transendothelial migration, de-coating into constituent parts occurs together with epithelial-mesenchymal transition (EMT), mesenchymal-epithelial transition (MET), mesenchymal-mesenchymal plasticity (MMP), recruitment of immunocompetent cells, senescence, and dormancy. Fig. 2. Metastatic niche 1 (MN-1) transforms into Metastatic niche 2 (MN-2): This transformation is induced by chronic inflammation, and remodeling through LOX with increased stiffness and decreased elasticity. LOX leads to increases in lamellipodia/motility. Recruited macrophages, platelets, and neutrophils lead to pro-tumorigenic TACs: TAMs (M2), TAPs, and TANs. CAFs secrete EVs and CCL2, thus leading to mesenchymal stem cells (MSCs), and immune suppression via decreased CD8+ T-cells and increased CD4+ TReg cells. These changes in the microenvironment result in consistent increases in granulosin, periostin, CXCL12, fibronectin, and K19, which further increase motility, and promote the migration of non-dormant CAFs and cancer cells. Fig. 3. Metastatic niche 3 (MN-3) fulfills the predisposing conditions for metastases or triggers dormancy: Increased cell plasticity induces active elongated cancer cells and newly developed metastasis-associated fibroblasts (MAFs), thus re-triggering dormancy. Anticancer therapy induces dormancy awakening, gelsolin with atypical EMT to spherical non-active cancer cells. Increased heterogeneity leads to formation of metastatic cancer satellites and immune suppression: (1) metastatic cancer cells and (2) MAFs migrate along gradients; (3) metastatic cancer cells are surrounded by MAFs; (4) CXCL12 and Keratin 19 coat metastatic cancer cells; (5) platelets surround metastatic cancer cells and (6) MAFs; and (7) neutrophil extracellular traps (NETs) shield metastatic cancer cells and (8) MAFs. Summary: Distant metastatic niches create high heterogeneity in CAFs, MAFs, and dormant and awakened cancer cells, which progress to form metastatic cancer satellites that fulfill the predisposing conditions for metastases.
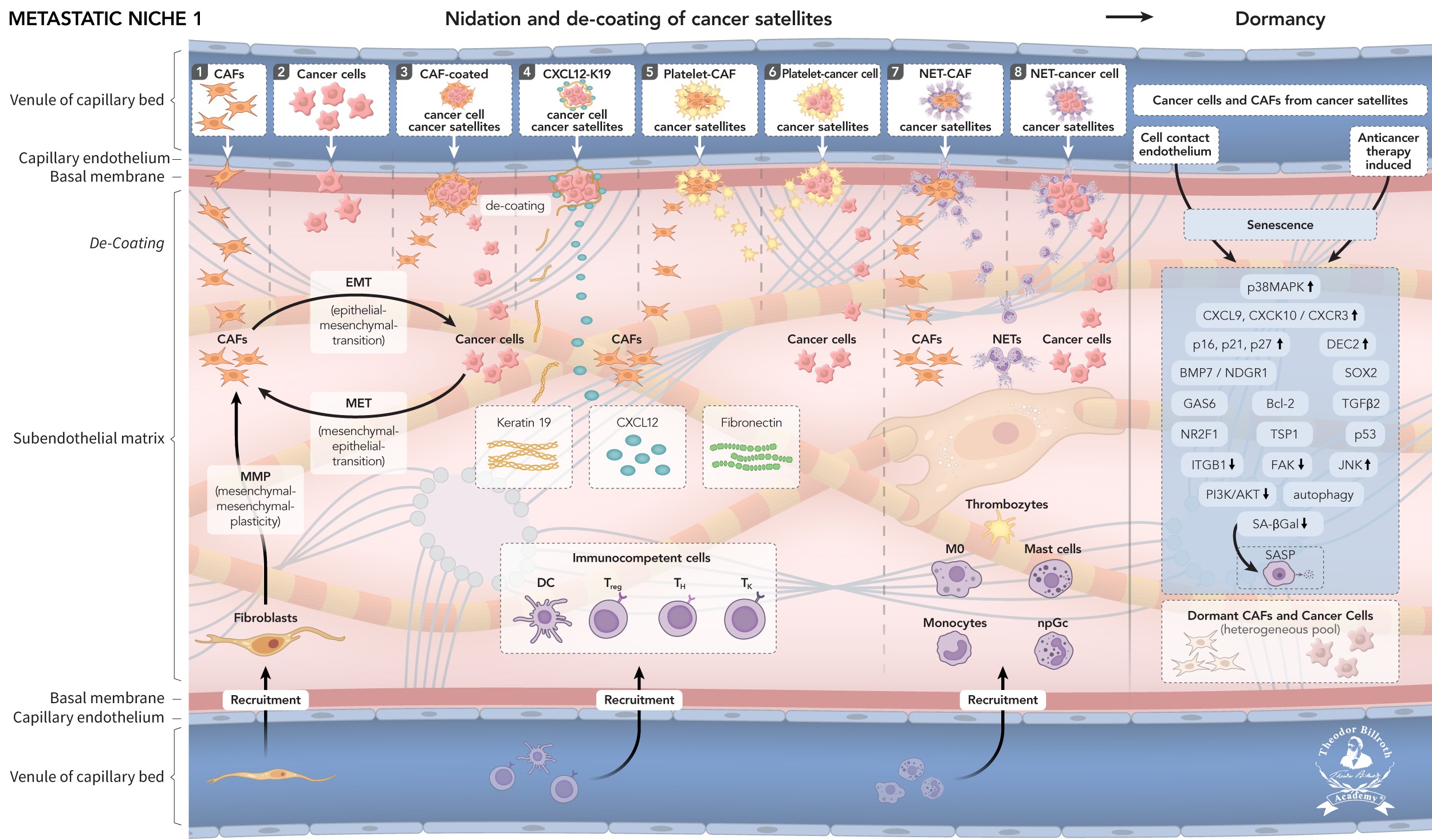
Fig. 1: Fig. 1. Metastatic niche 1 (MN-1): Pre-metastatic niche 3 (PMN-3) 39, according to current knowledge, results in traveling of a series of eight heterogeneous cancer satellites develop, including Trojan horses (immune evasion), alongside reciprocally affecting sequences, and subsequently travel (or, as usually described, “circulate”) alone or in combination, far from the primary tumor: (1) Cancer cells and (2) CAFs both migrate along the CXCL12 and fibronectin gradient. (3) CAFs surround cancer cells and migrate. (4) CXCL12 and K19 coated cancer cells migrate. (5) CAFs and (6) cancer cells are surrounded by platelets and migrate. (7) Neutrophils form neutrophil extracellular traps (NETs) that shield CAFs and (8) cancer cells. These cancer satellites extravasate past traveling, and nidate at the capillary bed. After transendothelial migration, de-coating into constituent parts occurs, together with cell epithelial-mesenchymal transition (EMT), mesenchymal-epithelial transition (MET), and mesenchymal-mesenchymal plasticity (MMP), which further increase heterogeneity; recruitment of immunocompetent cells; senescence; and dormancy. In addition, because of endothelial cell contact, as well as applied anticancer therapy, heterogeneous subendothelial cells transform through senescence into dormancy; this transformation occurs through a complex signaling and crosstalk network involving p38MAPK, CXCL9, CXCL10/CXCR3, p16, p21, p27, DEC2, BMP7/NDGR1, SOX2, GAS6, Bcl-2, TGF, NR2F1, TSP1, p53, ITGB1, FAK, JNK, PI3K/AKT, autophagy, and dormancy.
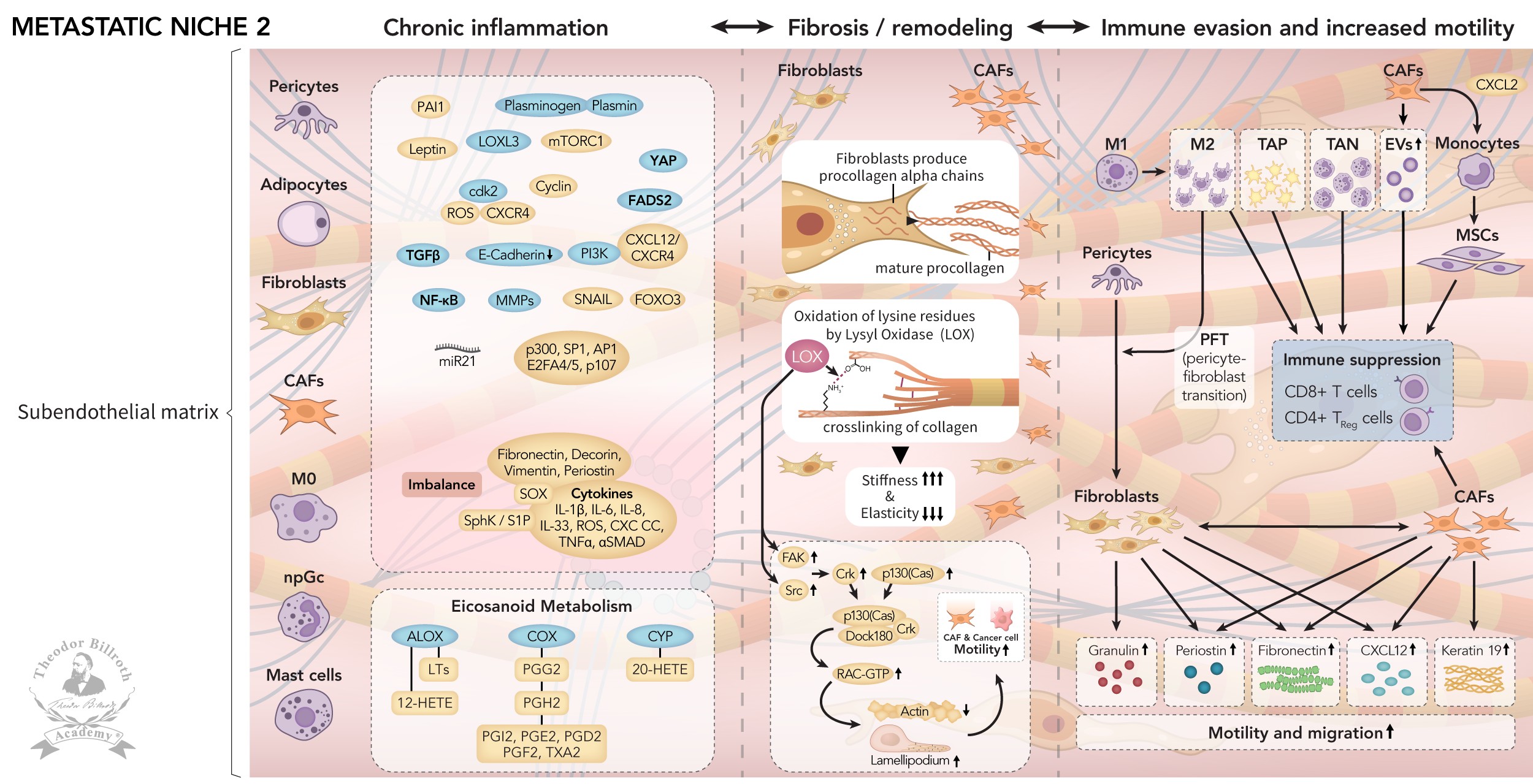
Fig. 2: Fig. 2. Metastatic niche 2 (MN-2): Ongoing signaling and crosstalk increase recruitment of pericytes, adipocytes, fibroblasts, CAFs, macrophages, neutrophils, and mast cell induced complex chronic inflammation via TGF, E-cadherin, NF-kB, PI3K, Snail, FOXO3, CXCL12)/CXCR4, PAI1, mTORC1, ROS, ck2, YAP, FADS2, miR21, p300, SP1, AP1, E2FA4/5, p107, fibronectin, decorin, vimentin, periostin, Sox, and cytokines (such as IL-1β, IL-6, IL8, IL33, ROS, IFNγ, TNFα, and αSMAD). This process includes activation of eicosanoid metabolism via CYP, COX, and ALOX to 20-HETE, PPG2, PGH2, PGI2, PGE2, PGD2, PGF2, TXA2, LTs, and 12-HETE. The result is fibrosis with collagen crosslinking through oxidation of lysine residues (remodeling) via lysyl oxidase, followed by increased stiffness and decreased elasticity. This leads again to FAK, p130 (cas)/crk/DOCK180 formation, with increased lamellipodia/motility. CAFs secernate extravesicular vesicles (EVs) and CCL2, with monocyte transformation into mesenchymal stem cells (MSCs). Each, together with pro-tumorigenic tumor-associated cells (TACs), e.g., TAMs (M2), TAPs, and TANs, results in immune suppression, through decreased CD8+ T-cells, and increased CD4+ TReg cells. Parallel increases in granulosin, periostin, CXCL12, fibronectin, and K19 further increase motility and migration.
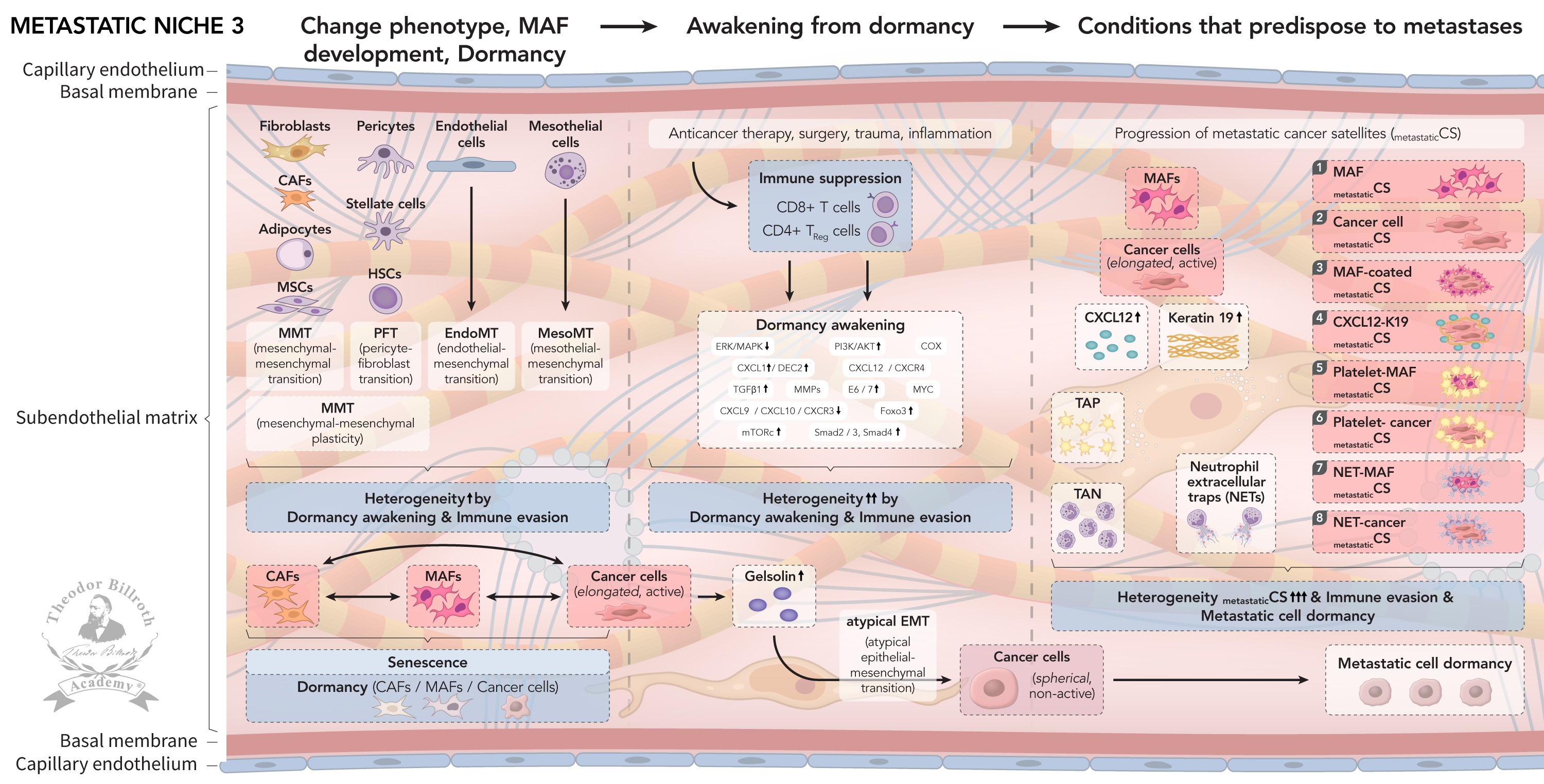
Fig. 3: Fig. 3. Metastatic niche 3 (MN-3) involves increased plasticity with phenotype changes through mesenchymal-mesenchymal plasticity (MMP) via mesenchymal-mesenchymal transition (MMT), pericyte-fibroblast transition (PFT), endothelial-mesenchymal transition (EndoMT), and mesothelial-mesenchymal transition (MesoMT). These processes substantially increase heterogeneity, as well as CAFs’ transition to MAFs and active elongated cancer cells, and again dormancy. Induced immune suppression, with decreases in CD8+ T-cells, and increases in CD4+ TReg cells, together with anticancer therapy, surgery, trauma, or inflammation, results in re-wakening from dormancy, followed again by increased heterogeneity. Increased gelsolin, through atypical epithelial-mesenchymal transition (atypical EMT), results in spherical non-active cancer cells. Finally, the conditions together with MAFs, cancer cells, CXCL12, K19, TAPs, TANs, and NETs are fulfilled for eight heterogeneous metastatic cancer satellites to develop, including Trojan horses (immune evasion): (1) metastatic cancer cells and (2) MAFs both migrate along the CXCL12 and fibronectin gradient. (3) MAFs surround metastatic cancer cells and migrate. (4) CXCL12 and K19 coated metastatic cancer cells migrate. (5) MAFs and (6) metastatic cancer cells are surrounded by platelets and migrate. (7) Neutrophils form neutrophil extracellular traps (NETs) that shield MAFs and (8) metastatic cancer cells. Consequently, increases in immune evasion and heterogeneity occur via metastatic cancer satellite dormancy.
Introduction
Living conditions in the Western world have improved, as reflected by an increase in life expectancy from approximately 40 years in the 1800s to approximately 60 years in the 1900s and approximately 80 years at present [1]. The manifold reasons for this include decreased infant and child mortality; improved hygiene, changes in work environments and fewer hazardous conditions, improved nutrition, and successful acute infection control through antibiotics, vaccination, and broad acceptance of scientific principles. The causes of death have shifted by improved medicine to chronic diseases, by enhancing the understanding, diagnosis, and treatment of chronic diseases, including cardiovascular diseases, diabetes, and, to a much lesser extent, cancer. Cancer is the second leading cause of morbidity and mortality worldwide after infectious diseases. The greatest successes in treating cancer are achieved when the cause of cancer can be identified; examples include human papillomavirus-induced cervical cancer and oropharyngeal cancers, and hepatitis C-induced liver cancer. Further major accomplishments have been made against leukemia, lymphoma, myeloma, melanoma, and pediatric cancers, which make up less than 10% of all cancers. Epithelial cancers account for approximately 80% of all cancers, and the success of their treatment, although heterogeneous, is measured by median survival of only weeks or months [2]. A recent meta-analysis examining the estimated life expectancy gains through cancer prevention, on the basis of 18 long-term randomized clinical trials involving 2.1 million individuals, has reported modest results, with little lifetime gain of days, with interventions such as sigmoidoscopy (110 days) mammography (0 days), prostate cancer screening (37 days), colonoscopy (37 days), fecal occult blood testing (0 days), and lung cancer screening (107 days) [3]. A study involving a randomized once-only sigmoidoscopy, with or without one fecal immunochemical test, or no screening in patients 50–64 years of age without colorectal carcinoma (CRC) at randomization, has indicated a cumulative 23-year CRC risk of 4.3% in the screened group and 6% in the unscreened group; adding fecal occult blood testing did not change screening benefits; otherwise, offering sigmoidoscopy decreased CRC risk by 1.7% (95% CI, −2.2 to −1.2 percentage points) [4]. Furthermore, CRC is the leading cause of cancer mortality in patients younger than 50 years [5]. These findings, together with markedly rising cancer incidence and nearly unchanged rates of metastases, illustrate the need to acknowledge that primary questions regarding the causal origins of cancer and metastases in epithelial cancers which have been ignored for too long. Altogether, improvements in living conditions and their beneficial effects on increasing life expectancy should not be misinterpreted to suggest that the gains in life expectancy are due to better cancer therapies. Although adjusted epidemiological cancer mortality rates are helpful for comparison between patient groups in different regions or countries, they can provide a false sense of comfort [2]. Currently, age-adjusted cancer data are often touted as evidence of successes against cancer. However, age-adjusted cancer data should not be mistaken for precise measures, particularly when demographic changes occur over long periods, in which ages and outcomes do not remain consistent. Notably, when demographic shifts occur over long periods (e.g., in the current aging population), adjusted rates have diminished reliability, because underlying changes might potentially be masked. Therefore, age-adjusted data provide only a relative and hypothetical index, but are not an actual and precise measure. Consequently, considering age-adjusted data as evidence of success in cancer is not always as fruitful. The unadjusted cancer mortality rates in Germany and the USA over the past 80 years have remained relatively unchanged [2]. Genetics alone does not reliably predict most common diseases, mortality, or disease progression. Its predictive value is strongest in monogenic disorders, whereas most complex diseases arise from interactions between genetic susceptibility, environmental exposures, physiology, and stochastic processes. Biomarkers are not always reliable, and mismatch repair gene defects have clinical support in approximately 10%–15% of epithelial cancers, but not in the vast majority [6–28]. Although biomarkers are measurable, they do not necessarily indicate the presence of a disease, because nature maintains living systems in homeostatic balance. A measured biomarker can be completely unimportant in maintaining homeostasis and can contradict its function. Reducing a disease to one signal (i.e., a biomarker) may be inappropriate if the context of its function in signaling, and crosstalk is not also identified. For example, a genome-wide association study in 124, 682 individuals across eight longitudinal studies (FinnGen, UK Biobank, Estonian Biobank, Generation Scotland, Genomics England, Genes & Health, Dana–Farber Cancer Institute, and BioMe) examined nine diseases (CRC, prostate cancer, breast cancer, coronary artery disease, heart failure, type 2 diabetes, chronic kidney disease, Alzheimer disease, and stroke). Moreover, biomarkers, with the exception of those for certain rare cancers, are not reliable. Furthermore, twin studies were believed to be able to provide evidence of the genetic basis of cancers because monozygotic twins are genetically identical, whereas dizygotic twins share 50% of their genes. An analysis of 44, 788 pairs of twins, one with and one without cancer in Swedish, Danish, and Finnish registries has assessed cancer risk at 28 anatomical sites. The twins without cancer had only a moderate absolute risk of having cancer at the same site as their twin with cancer [29]. Importantly, successes in only 10% of cancers, such as leukemia, lymphoma, myeloma, and melanoma, cannot and should not be extrapolated to most epithelial cancers, which have completely different tumor biology and conditions for disease progression and metastasis. During a 25-year timeframe, the fundamentals of the origin of carcinogenesis and metastasis, titled Epistemology of the Origin of Cancer, were addressed. Part I, 2014–2022, discussed how carcinogenesis develops [16, 17, 20, 21, 30–35]. Most epithelial cancers develop in response to (1) a pathogenic stimulus that induces (2) chronic inflammation, which is followed by (3) fibrosis with remodeling, from which (4) a precancerous niche (PCN) develops. This sequence of events was later independently demonstrated in an animal model [36]. Part II, 2023, addressed which is the first cancer cell [37]. Our model suggests that fibroblasts may be the first cells to undergo neoplastic transformation. The precancerous niche (PCN) induces (5) a chronic stress escape strategy (CSES), including epithelial-mesenchymal transition (EMT) in epithelial cells, which leads to the differentiation of CAFs expressing both epithelial and mesenchymal markers; finally, (6) CAFs undergo mesenchymal-epithelial transition (MET), and epithelial markers facilitate integration into the epithelium. Fibroblasts are the initial precursors of cancer, as first stated on Apr 27, 1858, by Rudolf Ludwig Karl Virchow (1821-1902) [38]. Part III, 2025, addressed how metastasis arises [39]. The Recamier-Fuchs-Paget seed and soil theory [40–42] cannot explain metastasis, because only 0.2% of injected cancer cells survive after 2 weeks in the lung [43, 44], and 99.8% of cancer cells are effectively eliminated by the immune system. The circulating tumor cell (CTC) theory [45, 46] is not only incomplete but also cannot explain metastases. CTCs are not synonymous with metastases, because no characteristics of cancer cells predict aspects including metastatic potential, growth rate, and chromosome numbers; moreover, even highly metastatic cell clones do not necessarily grow faster than normal cells [47]. We have proposed that metastasis occurs in parallel with carcinogenesis after the PCN is first transformed into three premetastatic niches (PMNs) [39]. Here, we suggested the concept of cancer satellites in PMNs defined as: a cancer satellite is a cell or particle escorting another from one location to another more distant location [39]. We suggest, this applies to the creation of metastatic niches (MNs) as well, although there is still a knowledge gap, as some cancer satellites still require scientific verification. Otherwise, some cancer satellites had already been observed previously, but were not interpreted as such as in the manner proposed earlier [39], or here: platelets surround cancer cells followed by immune evasion [48, 49], and aggregate with cancer cells followed by immune evasion [50, 51]; anti-platelet aggregation by aspirin [52] or inhibition [53, 54] results into favorable prognosis and diminished clinically observed metastases; neutrophil extracellular traps (NETs) from tumor-associated neutrophils (TANs) wrap around cancer cells and protect such against immune attack [55]; CXCL12–Keratin-19 filamentous formation can coat cancer cells with immune evasion [56]; CAFs coat cancer cells and suppress T-cell infiltration by CXCL12 [57]. In pre-metastatic niche 1 (PMN-1), lysyl oxidase (LOX) induces focal adhesion kinase (FAK) and breast cancer anti-estrogen resistance protein 1 (p130(cas)/adapter protein (crk)/dedicator of cytokinesis (DOCK180) formation, thus increasing lamellipodia and cell motility. CAFs and cancer cells secrete fibronectin and the chemokine C-X-C motif chemokine 12 (CXCL12, stromal cell-derived factor 1, SDF-1). Cancer cells also secrete Keratin 19 (K19), a prerequisite for increased migration. In parallel, platelets, neutrophils, and macrophages are recruited to PMN-1. One important unanswered question pertains to the conditions necessary for metastasis to develop. Answering this question is complex, because the reason why greater heterogeneity is observed in metastasis than in the primary cancer must be explained in both clinical and molecular biological terms. The transformation from PMN-1 to pre-metastatic niche 2 (PMN-2) involves tumor associated cells (TACs) from tumor associated macrophages (TAMs), tumor associated platelets (TAPs), and tumor associated neutrophils (TANs), thus leading to the transformation of anti-tumorigenic TACs into pro-tumorigenic TACs, local immunosuppression, a CXCL12 and fibronectin gradient, formation of lamellipodia, blebbing, and formation of neutrophil extravesicular vesicles (EVs), which together facilitate the mobility of CAFs and cancer cells, and allow them to migrate toward the endothelium. PMN-2 transforms into pre-metastatic niche 3 (PMN-3), which serves as a prerequisite for metastasis. A series of eight heterogeneous cancer satellites develop, including Trojan horses (which facilitate immune evasion) and reciprocally affecting sequences that travel alone or in combination [illustration 3 in 39]. These satellites move along the fibronectin- and CXCL12 gradient, (1) cancer cells and (2) CAFs travel; (3) CAFs surround cancer cells and travel along the CXCL12- and K19 gradient; (4) CXCL12-K19-coated cancer cells migrate; platelets (5) surround CAFs and (6) cancer cells; and neutrophils form neutrophil extracellular traps (NETs), which (7) shield CAFs from immune attack and (8) allow cancer cells to migrate. This sequence explains three important observations: (1) the logical associations found in patients with cancer after blood transfusion, (2) decreased disease-free time and survival, and (3) elevated metastasis rates, particularly in the context of TACs, because of transfusion-induced activation of TACs, particularly by TAPs. The current multimodal approach to cancer therapy can now be seen in a new previously unrecognized light. The benefits of such standard therapies might rely not on direct cancer cell effects but on indirect cytopenic effects, which have been regarded as merely adverse effects but can now be understood to interfere with the anticancer function of platelets, macrophages, and neutrophils, and to further exacerbate disease progression and metastasis. To gain a comprehensive understanding, closer examination of aspects of extravasation, such as capillaries, adhesion, soluble selectins, pericytes, and transendothelial migration, is necessary.
Extravasation, capillaries, and transendothelial migration
Readers are referred to the Supplemental materials for details regarding many aspects of signaling and crosstalk complexity and heterogeneity.
Extravasation Extravasation is the exit of cancer cells from the vascular system at distant sites from the primary tumor location. Blood arrives at distant capillaries and consists of water; cell components; and plasma components such as albumin, fibrinogen, coagulation factors, globulins, electrolytes, nutrients (e.g., amino acids and lipids), hormones, metabolic waste, and oxygen. Blood is also the transport medium for other cells, such as CAFs, cancer satellites, and cancer cells, which have been observed by intravital videomicroscopy to arrive at the sinusoids of the liver and to arrest in narrow capillaries [58]. Platelet endothelial cell adhesion molecule (PECAM-1, platelet endothelial cell adhesion molecule, CD31) is found on many cells, such as endothelial cells, platelets, macrophages, lymphocytes, and Kupffer cells [59. In an experimental model of hepatic metastasis based on splenic injection of CRC cells, the sinusoid location of Kupffer cells, fibroblasts, and PECAM-1 were revealed [60].
Capillaries Capillaries consist of endothelial cells (in a single flat cell layer), a layer of connective tissue (basement membrane), and pericytes [61, 62]. Pre-capillary arterioles consist of the tunica intima, tunica media, and tunica adventitia, which post-capillary venules that consist of most endothelial cells. Capillaries in the human body consist of (1) non-fenestrated capillaries (skin, lung, brain), (2) continuous fenestrated capillaries (intestines), and (3) discontinuous capillaries (sinusoids) (liver and bone). Before adhesion or transendothelial migration, cell-cell communication is necessary (Supplement, part 1, Capillary signaling and crosstalk). Selectins have previously been reviewed in detail [39]. Here, we discuss soluble selectins, which are important for adhesion and nidation (Supplement, part 2, Soluble selectins). In this regard, phagocytic, contractile, and pluripotential pericytes function as sentinels of the distant tumor microenvironment (Supplement, part 3, Pericytes). After adhesion and nidation, transendothelial migration occurs.
Transendothelial migration The transendothelial travel of immune cells occurs very rapidly. Laminin 511 is a bouncer, in that it stabilizes cadherin in between endothelial cells through β1 and β3 integrins, in a manner mediated by RhoA signaling [63], and downregulates the transmembrane protein CD99 molecule like 2 (CD99L2), which is a member of the Ras homolog family member A (RhoA) proteins responsible for decreasing neutrophil extravasation [64]. Notably, immune cells require approximately 40 minutes to travel through the basal membrane but only 2 minutes to pass through the endothelium. Perspectives regarding various cell compartments must be viewed in the context of their cell turnover rate.
Cell turnover A 70 kg human has an estimated 30 trillion cells (30, 000, 000, 000, 000) [65], with a turnover rate of 0.33 ± 0.02 × 1012 (330 ± 20 billion) cells d−1, which is equal to approximately 4 million cells s−1, although cell lifespan markedly varies by type and tissue [66]. Approximately 86% of total cell number and turnover consists of blood cells (65% erythrocytes, 18% neutrophils, 12% gastrointestinal epithelial cells, 2.2% lymphocytes, 1% skin cells, 0.5% monocytes, and 0.1% endothelial and lung cells). Additionally, ~ 38 ± 10 trillion bacteria reside in the colon. The neutrophil turnover rate, according to an average of two estimates (bone marrow and the circulating pool) is approximately 6 ± 1 × 1010 cells d−1; therefore, ~60 billion cells (60, 000, 000, 000 cells) are produced every 24 hours. In contrast, CTCs travel very inefficiently, because only 0.2% become metastatic [43]; therefore, the immune system is highly effective in attenuating 99.8% of CTCs in the bloodstream [39]. An incompletely understood aspect of metastases is the heterogeneity of cancers and metastases, including micro-metastases (Supplement, part 4, Heterogeneity). Other important variables include senescence, dormancy, and reawakening from dormancy. The interplay between dormancy and reawakening not only increases cell plasticity but also increases cell heterogeneity. No one variable can distinguish which cell derives from which intermediate stage of senescence, dormancy, or reawakening, and why. Plant biology has demonstrated that senescence, dormancy, and reawakening are fundamental physiological processes in a strategy for survival in unfavorable environmental conditions. This principle also governs metastasis, because dormancy occurs only under special circumstances with varying durations between nidation, colonization, and reawakening.
Dormancy
The dormancy concept was described in 1867 by Charles Moore (1821–1870), who stated that a “residual fragment of the original disease should remain quiescent for years” [67]. In 1948, the heterogeneity in short- versus long relapse-free intervals (e.g., periods of months in gastric and colon carcinoma, approximately 1 year in kidney or tongue carcinoma, and longer periods of quiescence in melanoma and breast cancer) was reported by Rupert Allan Willis (1898–1980) [68]. Geoffrey Hadfield (1889–1968) examined the high variability in the interval between cancer surgery and the appearance of relapsed cancer and coined the term “dormant cancer cell.” He stated, “When the interval is prolonged to six years or more it seems impossible to escape the conclusion that the cells of the dormant growth are in a state of temporary mitotic arrest, no matter how long the period may be” [69]. He further used dormancy “to describe malignant cells which, although remaining alive in the tissues for relatively long periods, show no evidence of multiplication during this time, yet retain all their former and vigorous capacity to multiply.” According to Hadfield, dormant cells were located near the tumor’s surgical site within the scar, in poorly vascularized mature collagenous connective tissue with relative anoxia or in regional lymph nodes. He assumed that mitotic arrest could be induced by anoxia and that reactivation occurs after oxygen becomes available again (through re-vascularization or inflammation). Cells in non-proliferating stages can be in quiescence, dormancy, or senescence [70]. Senescent cells are metabolically active but non-proliferative [71]. Quiescence (or latency) (Latin: quietus, at rest) is a reversible resting state involving cell division and growth that are repressed under an unsuitable microenvironment but can be reactivated under favorable conditions [72, 73]. An example from botany is the removal of cellular water in plants through the accumulation of protective molecules and formation of intracellular glasses; after rehydration, and in the presence of suitable temperature, light, and oxygen, the plants resume metabolism and germinate [74]. Dormancy (Latin: dormire, to sleep) is defined as a reversible state of metabolic activity with reversible proliferation arrest occurring in the G0 or G1 phase of the cell cycle (arrested growth), which can be reactivated under proper conditions in the presence of a necessary specific trigger [75–77]. Quiescent cells have been proposed to be in G0 stage, whereas dormancy would constitute a deeper cell cycle arrest [74], and cells arrested in G1 phase are often quiescent [78, 79], thus potentially explaining why the terms quiescent and dormant are frequently used interchangeably. Dormancy, a reversible state of diminished metabolic activity, has been observed in viruses, bacteria, fungi, protists, worms, insects, crustaceans, amphibians, fish, birds, plants, and mammals. Dormancy is assumed to have contributed to the emergence of life on Earth, by not only ensuring the biodiversity of life but also decreasing the probability of extinction under limited resource conditions by providing reserve growth potential [80, 81]. A long time ago, it was stated “Dormancy of the seed during development is so general that it may almost be taken for granted” [82]. However, dormancy can occur at primary tumors as well as at distant sites of metastasis [83, 84]. Senescence (Latin: senescere, to grow old) is defined as terminal growth arrest that leads to death [85]; however, re-entry into a proliferative stage after p53 and p16 inactivation has been reported and termed reversible senescence [86, 87]. The G2-arrested quiescence in Drosophila neural stem cells has been reported as a reversible cell cycle arrest in the G2 phase [88]. Senescence differs from dormancy in that senescent cells are believed to undergo irreversible growth arrest associated with senescence-associated β-galactosidase (SAβ-gal) [89] and SA-β-gal accumulation [90], both of which are induced by suppressor cyclin-dependent kinase inhibitor 2A (p16, p16INK4a) [91], induction of human chromosome 7, or segments of an in vitro established human fibroblast line (SUSM-1) [92]. Lysosomal galactosidase beta 1 (GLB1) [93] encodes an SAβ-gal, an extrinsic peripheral membrane protein, that serves as an elastin receptor [94] in senescent cells. The period from the presence of dormant cancer cells and cell complexes/satellites in the stroma to the emergence from dormancy with metastatic progress has been termed metastatic cancer dormancy [76]. In breast cancer, long latencies of 10–15 years between treatment and relapse, even in node-negative patients, are frequently seen [95, 96]. Long latencies are also seen in node-negative early breast cancer, which increases in dependency of Her2/neu, triple negativity, and estrogen receptor positive (ER+) cohorts [97]. More broadly, dormancy should be considered a normal phenomenon rather than a pathological condition. Cluster of differentiation 154 (CD154), which is expressed on CD4+ T-cells, CD8+ T-cells, macrophages, natural killer (NK) cells, mast cells, platelets, endothelial cells, and epithelial cells, binds CD4+ T cells, thus forming cluster of differentiation 40 (CD40). Signaling and crosstalk result in CD40 activation and initiation of the senescence-associated secretory phenotype (SASP) in non-small-cell lung cancer [98]. Notably, senescent cells secrete inflammatory cytokines plus extracellular matrix degrading proteins participating in the remodeling of the extracellular matrix. Dormant and quiescent cells have been suggested to re-enter the cell cycle and proliferate [78]. Dormancy of occult cancer cells is believed to indicate metastatic potential. “Chemotherapy is not effective in purging bone marrow even in chemo-responsive patients” [99]. Combining high dose chemotherapy with autologous bone marrow transplantation has revealed effective cancer cell elimination, although combinations of various antibodies have not demonstrated enhanced effectiveness [100]. Importantly, most malignant cells and their complexes are in different stages after traveling to distant sites [101–103], thus potentially explaining their relative susceptibility to chemotherapy. The final step between dormancy and reawakening in metastatic cancer is the exit from quiescence into cell-cycle re-entry, in a process often described as “metastatic reawakening” or dormant cell reactivation. In the literature, this transition is frequently associated with a shift in signaling balance away from dormancy programs, such as p38, and toward pro-growth extracellular signal-regulated kinase (ERK) signaling, thus allowing disseminated tumor cells to proliferate again. Dormant disseminated tumor cells can persist in a non-proliferative state for years, held in check by signals from the tumor niche, immune surveillance, and extracellular matrix cues. When these constraints are lifted, the cells can re-enter the cell cycle, adapt to the new tissue environment, and progress to metastatic growth. Mechanistically, this process can be explained by dormancy maintenance due to release-from-dormancy signals, cell-cycle re-entry, clonal expansion, and detectable metastatic relapse [104–106]. Currently, dormancy is differentiated into angiogenic [107], immunomediated [108–111], and cellular dormancy, and proliferation is balanced against cell death [112]. Furthermore, dormancy is now known to result from decreased proliferation and not increased apoptosis [113]. This non-proliferative arrest is both reversible and resistant to multimodal cancer therapy. Dormancy is associated with protein kinase C gamma gene (PRKCG) signaling and increased stromal remodeling [114], as well as the (ERK)(MAPK)/p38(SAPK) activity ratio [115], because the tumor suppressor p38 induces G0/G1 arrest and dormancy [116–119]. Single cancer cells are usually dormant, specific tissue environments favoring ongoing growth and nidation [120, 121]. In such dormant cells, chemotherapy is ineffective [122]. M2 (pro-tumorigenic) macrophages can induce dormancy by increasing angiostatin [123] and by inhibiting neovascularization in cancer, thus resulting in dormancy [124]. Notably, aspirin can upregulate uPAR in human colon cancer [125], and inhibition of uPAR, β1-integrins, FAK, or eGFR, alone or in combination, can result in dormancy in vivo [126]. Reversal from dormancy to fast growing cancer has been observed in various epithelial cancers [127–129]. Reawakening from dormancy is a natural process involving altered stromal cues, decreased dormancy-inducing factors such as secreted protein, transforming growth factor beta2 (TGFβ2) in some niches, restored ERK activity, and integrin/FAK signaling, all of which promote proliferation. No precise reactivation sequence for dormancy awakening exists because disruption of homeostasis must first occur. An increase in the ERK/p38 MAPK activity ratio with a shift from p38HIGH/ERKLOW dormancy to ERKHIGH/p38LOW drives re-proliferation [76, 115]. Next, because of angiogenesis induction [130], even the fibronectin production in the cytoskeleton can result in awakening [131]. Inflammation [132] and chemotherapy also induce awakening [133–135]. Awakening can be triggered by chemotherapy NET induction [134]. A recent paper has attracted attention by reporting that Rous sarcoma virus infection promotes breast cancer cell awakening [136]. Cortisol, epinephrine, norepinephrine, and serotonin activate neutrophils; subsequently, inflammation increases via S100A8 and S100A9, thus altering the distribution of lipids, which accumulate in neutrophils, and leading to reactivation of dormant cells [137]. Because cyclooxygenase (COX) itself can result in awakening, COX2 inhibitors can prevent reactivation [138]. During dormancy, the signaling pathways of phosphoinositide 3-kinase (PI3K)-AKT and mitogen-activated protein kinase (MAPK), together with RAS–MEK–ERK, are suppressed [139, 140]. Cancer cells themselves can induce PI3K together with autophagy induction by EGFR phosphorylation, AKT inhibition and by inducing Cyclin D1 [139]. Bone stromal cells secrete the TGF-β family member bone morphogenetic protein 7 (BMP7), which, in a BMP receptor 2 (BMPR2) dependent manner, activates p38 MAPK followed by the cell cycle inhibitor p21, and the metastasis suppressor gene N-myc downstream-regulated gene 1 (NDRG1) with cell cycle arrest [141]. Withdrawal of BMP7 restarts cell growth, whereas application of BMP7 suppresses growth, thus explaining why the interplay of BMP7–BMPR2–p38–NDRG1 signaling directly influences dormancy versus awakening, and that why senescence is not irreversible (as previously assumed). Even inactivation of the tumor repressor p53 reverses senescence in fibroblasts [86]. TGFb2 Smad1 signaling induces the metastasis suppressor DEC2 and BMP7 [141]. CAFs have been shown to secrete CXCL1 with downregulation of DEC2 promoting awakening of dormant cancer cells [142]. CXCL12 release by hepatic stellate cells (HSCs) promote through CXCR4 to exit quiescence and induce cell growth [143]. CXCL9 keeps dormancy stable [144], as does CXCL10/CXCR3 signaling [145], whereas its inhibition results in awakening. Dormancy can be maintained by TGFb through Smad complexes increasing p27 and p21 [146]. Smad4 is essential for transnuclear Smad2/3 transportation and is also a tumor suppressor; notably, Smad4 knockdown reverses p27 activation and growth repression in epithelial cancer cells [147]. Although inactivation of the oncogene MYC results in a dormant state, its reactivation results in awakening [105], as do AP-1 and NF-kB [148, 149]. Matrix metalloproteinase 9 (MMP9) and neutrophil elastase released by NETs induce 𝛼3 𝛽1signaling and awakening of dormant cancer cells [150]. Reoxygenation also results in expression of the E6 and E7 oncogene (E6/7), awakening, and proliferation [151]. E6/7 often result in deregulation of the transcription factor FOXO3 [30, 152]. FOXO3 keeps cells in dormancy via p12 and p27 [153], and PI3K/AKT signaling suppresses FOXO3a promoting cell growth [154]. FOXO3 is an inhibitor of the mammalian target of rapamycin complex 1 or mechanistic target of rapamycin complex 1 (mTORC) with activation of AKT [155]. Suppression of mTORC activity results in cell cycle arrest, including proteomic changes, whereas removal of mTOR inhibitors re-establishes cancer cell growth [156, 157]. Activation of transcription factor 6 alpha (ATF6α) upregulates the GTPase Ras homolog enriched in brain (Rheb), a critical AKT-independent mTOR signaling inducer [140]; ATF6α-Rheb-mTOR signaling enhances dormant cell survival in vivo, whereas deletion of mTOR in hematopoietic stem cells and progenitor cells (HSPCs) results in a loss of quiescence and increased proliferation [158]. Further information on dormancy is provided in the Supplement (Supplement, part 5, Dormancy). The interplay between inflammation and immune evasion with induction of a pro-metastatic microenvironment is important.
Chronic inflammation
Inflammation is a primary defense mechanism to eliminate harm and maintain homeostasis. Many cell types are involved, including fibroblasts and immunocompetent cells, such as antigen presenting dendritic cells (DCs), cytotoxic CD8+ killer T-cells, which require antigen recognition via MHC, natural killer T-cells (NK), which adapt rapidly without antigen specify, together with the coordinating CD4+ T-helper cells (TH), with various subsets Th1, Th2, Th17 and suppressing CD4+ regulatory T-cells (TReg), and mast cells, monocytes, macrophages, platelets, and recruited neutrophils. In chronic inflammation homeostasis is disrupted, inducing fibrosis and remodeling, through which the normal healthy state become a pro-tumorigenic and pro-metastatic milieu. In earlier publications, we discussed ongoing chronic inflammation in detail [16, 17, 30–34], including TGFb, decreased cell-cell-contact by CAM 120/80 (E-cadherin), and stimulated nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-kB), phosphatidylinositide 3-kinase (PI3K), with signaling disruption of metalloproteinases (MMPs), zinc finger protein SNAI1 (Snail), Forkhead box protein (FOXO3), CXCL12) and CXCR4 crosstalk. These interactions result in a persistent increase in chronic inflammation through plasminogen activator inhibitor-1 (PAI1), rapamycin complex 1 (mTORC1), reactive oxygen species (ROS), cyclin-dependent kinase 2 (ck2), yes-protein (YAP), and fatty acid desaturase 2 (FADS2, ω-6-desaturase (D6D)), with consequent effects on plasticity; microRNA-21 (miR21) triggered signaling increase in protein 300 (p300, p300-CBP coactivator family), specificity protein 1 (SP1), activator protein 1 (AP1), cytoplasmic complex of Smad3, E2F4/5 and D-prostanoid (DP1) (E2FA4/5), retinoblastoma-like protein 1 (p107, RBL1) and an imbalance of fibronectin, decorin, vimentin, periostin (POSTN), Sox and cytokines, such as interleukin 1 beta (IL-1β), IL-6, IL8, interleukin 33 (IL33), ROS, CXC CC, interferon gamma (IFNγ), TNFα, alpha-smooth muscle actin (αSMAD). These factors are each relevant, because their induction occurs at both distant sites and at the primary tumor. Furthermore, the activation of the NF-kB metabolism [32] is complex. In addition, eicosanoid metabolism [33] has complicated effects on cytochrome P450 isoforms (CYP), cyclooxygenase metabolism, and arachidonate lipoxygenase acitivity (ALOX), thus leading to the induction of various prostaglandins, prostacyclins, thromboxanes, and leukotrienes. Further information on the effects of chronic inflammation on metastasis signaling and crosstalk is provided in the Supplement (Supplement, part 6, Chronic inflammation). Chronic inflammation, which induces fibrosis and remodeling, further increases not only heterogeneity but also activation of TACs, including signaling and crosstalk. The roles of TACs in metastasis have recently been reviewed [39]. TACs comprise heterogeneous cell populations, including neutrophils, macrophages, and platelets, many of which have been implicated in key steps of the metastatic cascade. TACs are necessary for metastasis regarding neutrophils, TANs, and NETs including elastase, TAMs (M2) together with mesothelin and inhibin. TAPs and elastase are further discussed in the Supplement (Supplement, part 7, Tumor-associated cells). Direct or indirect suppression of the immune system results in immune evasion, an important mechanism during metastasis.
Immune evasion
Immune evasion occurs at various levels. Currently, successful organ transplantation is known to depend on genetic compatibility between the donor and host via the major histocompatibility complex (MHC), which also determines cancer transplantation. Furthermore, understanding of the roles of the MHC in cancer is important, because immunosuppression enables cancer growth. The clearance of cancer cells at distant sites occurs not only via the immune system but also independently at the endothelium by released nitric oxide in vivo [159]. Homeostasis is observed because NO derived from cancer cells can also have pro-tumorigenic effects [160]. The anti-tumorigenic immunoglobulin M has been found to increase metastasis in an experimental model [161]. Lamina propria macrophages express high levels of 25F9 (a marker for certain subpopulations of macrophages), MHC class II molecules, and CD74 (MHC class II-associated invariant chain) [162]. Cancer cells and CAFs downregulate MHC-1 class molecules, thus leading to immune evasion [163–165]. MHC-class recognition by cells influences the immune response. NK cells recognize MHC-I through specific receptors preventing them from elimination [166]. CAFs have relatively high mobility (via p130CAS, Src, Crk, RAC, and lamellipodia) but low proliferative ability [167]. High SMAD3, independently of TGFβ, recruits CAFs in lung adenocarcinoma. CAFs in turn increase MMPs [168] and downregulate MHC class I-restricted T-cells (CD8+ T-cells), which kill cancer cells [165]. TReg cells are also increased by CAFs [169], and CCL2 increases macrophage suppression [170]. Epithelial carcinomas rich in the endothelial apoptosis inducer Fas ligand (FasL) are associated with few CD8+ T-cells and a variety of regulatory T-cells. FasL inhibition results in tumor suppression [171]. Early B-cell factor 1 (EBF1) is important for B-cell differentiation in B-lymphocytes [172] and can initiate Cox-subunit 412 (COX4I2) transcription in CAFs, thereby promoting M2 macrophage activation and T-cell suppression in CRC [173]. Furthermore, EBF-1 binds CXCL12, Ccl9, and POSTN [174]. IL-10 producing regulatory B-cells (Bregs) [175] make up approximately 10% of B-lymphocytes [176] and regulate anticancer T-cell immunity; therefore, decreasing B-lymphocytes is a potential immunotherapy approach in cancer [177]. CD24highCD27+ B cells make up a high percentage of tumor-associated B cells in CRC tumors [178]. Decreasing peripheral CD19+CD24highCD27+ B lymphocytes in patients with gastric cancer treated with chemotherapy has been found to improve progression free survival [179]. CAFs induce chemokine (C-C motif) ligand 2 (CCL2) with monocyte recruitment and differentiation to mesenchymal stromal cells or myeloid-derived-suppressor cells, thereby suppressing the immune system and promoting angiogenesis [180–182]. CCL2 induces Smad3 Protein- and p42/44 mitogen-activated pProtein kinase (MAPK) signaling increases breast cancer motility [183]. CAFs modify M2-type macrophages, TReg cells, and myeloid-derived suppressor cells (MDSCs), thereby enhancing immune evasion [184, 185]. In parallel, ongoing chronic inflammation decreases host immunity and the anticancer T-cell response [186]. T cells and B cells, which largely direct the anti-tumor immune response, are notably diminished in number in metastases (only approximately 17%, in comparison to primary breast cancer, whereas the loss of immune activation is approximately 30%) [187]. Eliminating CD8+ T cells in cancer decreases survival [188], whereas the interaction of CD4+ plus CD8+ T-cells increases survival in esophageal cancer [189]. The regulation of CD8+ T-cell replication is increased by antigen presenting DCs [190]. However, various DCs exist and may have both anti- and pro-tumor effects. More details regarding immune evasion during PMNs have been provided in prior publications [39]. Details regarding immune suppression and evasion during metastasis are provided in the Supplement (Supplement, part 8, Immune evasion). The complex signaling and crosstalk of chronic inflammation, TACs, with immune evasion induce fibronectin, CXCL12, and K19, as previously reviewed in detail [ 39]. In cytoskeletal remodeling, gelsolin is important in enabling cancer cells to evade the immune system.
Gelsolin Gelsolin is a calcium-regulated actin depolymerizing protein involved in regulation of the cytoskeleton [191–193]. Gelsolin inhibits apoptosis and is cancer suppressive [194], and it is downregulated in cancer [195–198]. Gelsolin induces fibroblast and epithelial cell motility. TGFβ induces expression gelsolin, which in turn leads to the conversion of a migratory, stress-fiber-rich phenotype to a cortical actin-rich spheroidal state [199]. Simultaneously, biochemical stiffness and immune evasion decrease. Increased gelsolin results in atypical EMT from an elongated active cancer cell phenotype to a spherical non-active phenotype, which is considered a metastatic dormancy stage and therefore is not vulnerable to immune surveillance. Another induced secernated small peptide is granulin with its precursors.
Granulin Granulin and its progranulin form promote migration in breast cancer [200], and increase the H. pylori-induced proliferative activity and migration seen in gastric epithelium [201], in ovarian cancer with its CAFs [202], pancreatic cancer [203], gastric cancer [204], and CRC [205, 206]. Granulin increases migration of the highly suppressive subpopulation of CCR8+ TRegs in bone [207]. Cancer cells actively secrete K19, which prevents CXCL12 from protecting against immune attack [208]. K19 mRNA can be used to identify micro-metastasis in lymph nodes in patients with breast cancer [209]. K19 knockout decreases proliferation, whereas K19, together with Granulin, POSTN, fibronectin, and CXCL12, increases cancer cell proliferation [210]. Furthermore, K19 increases cell migration in pancreatic cancer via Wnt/β-catenin [211], although it inhibits cell invasiveness of primary breast cancer [212]. K19 directly promotes cancer cell survival, invasion, and angiogenesis, thus resulting in hepatocellular carcinoma (HCC) progression with poor clinical outcomes [213–215]. Another glycosylated protein important during metastasis signaling and crosstalk is POSTN.
Periostin (POSTN) POSTN (formerly known as osteoblast-specific factor 2) [216] is a 90 kDa glycosylated protein [217] with several isoforms [218]. This protein plays important roles in epithelial cancers, such as those of the breast [219], prostate [220], lung [221], esophagus [222, 223], stomach [219], ovary [219], head and neck [224], prostate [225], colon [225], colorectum with liver metastasis [225], and others [226]. POSTN is increasingly detected in lymph node, liver, bone, and lung metastases. The anti-fibrotic agent irfenidone inhibits pancreatic cancer desmoplasia; deposition of POSTN, fibronectin, COL1; and lymph node and liver metastases [227]. In bone, POSTN binds bone morphogenetic protein-1 (BMP-1), which in turn cleaves the LOX precursor propeptide [228]. Osteoblast-induced POSTN is elevated in bone metastases from prostate cancer [229], lung cancer [230], and breast cancer [231–234]. POSTN serves as a surrogate marker for bone metastases in breast cancer [235]. In the liver, POSTN is upregulated in CRC liver metastasis, induces COL1 and fibronectin via the TGFβ receptor, independently of Smad2/3 activation with LOX activation in HSCs promoting fibrosis [236, 237]. HSC-derived POSTN induces TGFβ in CRC cells through integrin/FAK/ERK/STAT3 and Smad, and its levels correlate with fibrotic features as well as TGFβ signaling in CRC liver metastases. Decreased POSTN results in decreased HSC activation and fewer liver metastases [238]. CAFs secrete EVs with activated LOX inducing remodeling and EMT through FAK/paxillin/YAP, interacting with POSTN, fibronectin, and bone morphogenetic protein-1 [239]. CAF EVs activate lung fibroblasts and promote matrix remodeling and POSTN release, together with increased fibronectin, LOX and MMP-9, increases in FAP, α-SMA and Ki67 participating in the creation of a metastatic niche in the lung [240]. POSTN promotes fibronectin expression/secretion and extracellular matrix deposition [241] and modulates myofibroblast differentiation via the integrinβ1/RhoA pathway and fibronectin synthesis [242]. POSTN-positive CAFs activate integrin-FAK/Src-vascular endothelial-cadherin (VE-cadherin) in lymphatic endothelial cells with enhanced metastatic dissemination [243]. Extracellular POSTN mediates fibronectin synthesis via integrin β1, FAK, and JNK [244], and directly binds fibronectin [217]. The membrane-bound mucin-like glycoprotein podoplanin (PDPN) also induces POSTN expression in fibroblasts and CAFs. More detailed information on PDPN is provided in the Supplement (Supplement, part 9, Podoplanin (PDPN)). The remodeling of the tumor microenvironment induced by chronic inflammation increases heterogeneity via cell transition plasticity.
Cell transition plasticity Another principle to help understand metastases is cell transition plasticity, which is essential for maintaining homeostasis and inducing cell and tissue regeneration, as well as for life. Plasticity is defined as reversible changes in phenotype, functional identity, or differentiation status without alteration of the genome. The high deformability (plasticity) of cancer cells correlates with the progression of the transformed phenotype from a nontumorigenic cell into a tumorigenic, metastatic cell [245]. Nearly 80 years ago, Conrad Hal Waddington (1905-1975) published the book “The Strategy of the Genesis,” which has provided the basis for epigenetics [246]. He described a paradigm for how hierarchical sequential terminal differentiation of cells leads to specialized cells within the development of an embryo from an egg. This paradigm was later modified by Sir John Betrand Gurdon (1933-2025), on the basis of nuclear transplantation and consequent formation of differentiated intestinal cells [247]. Shinya Yamanaka (1962) introduced transcription factors for development, and demonstrated that the differentiation of fibroblasts in mice, which was believed to be terminal at the time, could be reverted to a pluripotent stem cell state [248]. This field gave rise to epigenetics, which, along with Waddington’s concept of the epigenetic concept led to a persistent importance of the role of epigenetics in disease. Currently, increased cell plasticity is known to not only result from EMT and MET, but also to be enhanced by MMT and pericyte-fibroblast transition (PFT), both of which are representative of mesenchymal-mesenchymal plasticity (MMP), endothelial-mesenchymal transition (EndMT), or mesothelial-mesenchymal transition (MesoMT). Primary mesenchymal cells can induce E-cadherin activation with re-expression of apical-basal polarity, thereby inducing EMT [249]. Consequently, EMT with activation of TGFβ, NT-3, sonic hedgehog, and α5βl integrin occurs. Transfection of metastatic cells or normal embryonic fibroblasts with the E-cadherin gene has been found to convert normal cells into metastatic invasive cancer cells. Currently, this transdifferentiation is known to be a normal biological process that explains how hepatocytes morph into pancreatic ductal cells [250], how white adipocytes turn into brown adipocytes [251], and how endothelial cells morph into vascular smooth muscle cells [252]. EndMT was first discovered in the study of embryology in heart development [253], in microvessels [254], and in broader in vivo and in vitro studies in embryology [255]. Even cardiomyocytes can induce transdifferentiation from endothelial cells to cardiac muscle cells [256]. Shear stress can result in a phenotypic change from endothelial to epithelioid [257]. In injured vascular endothelium, bone marrow, monocytes morph into endothelial progenitor cells [258]. Therefore, why EndMT correlates with fibrosis-associated disease, ossification [259–262], cancer [259], malformation [263], and atherosclerosis [264] is currently understood. EndMT is a key link between chronic inflammation and endothelial dysfunction, and an early event in breast cancer metastasis, together with NO deficiency [265]. CAFs can be derived from the endothelium by EndMT [266]. In patients receiving peritoneal dialysis, mesothelial cells undergo MMT, a specific type of EMT, to a myofibroblast-like phenotype; HGF, TGFβ, Interleukin-1β, Vimentin, and fibronectin facilitate this process, thus resulting in extracellular matrix production and fibrosis [267–271]. These findings have been observed both in vitro and in vivo [272]. The phenomenon is associated with upregulation of thrombospondin-1 (TSP1), collagen-13 (COL13), and gremlin-1 (GREM1). It results in a decrease in epithelial markers such as E-cadherin and cytokeratin, and the induction of mesenchymal markers such as Snail, N-cadherin, fibronectin, collagen I, α-smooth-muscle actin (α-SMA), and fibroblast specific protein-1 (FSP-1). Mesothelial-to-mesenchymal transition (MMT) is a source of CAFs in peritoneal metastasis [273]. This process can be inhibited by PPAR-γ agonists or TGFβ inhibitors [274, 275]. LOXL2 triggered by chronic inflammation-induced TGFβ, which promotes EMT, and inhibition of LOXL2 to prevent EMT [276] and thus MMT. Pericytes (stellate cells) can differentiate into various cells including osteoblasts, phagocytes, adipocytes, and fibroblasts [277, 278]. In fibroblasts, matrix stiffness, together with YAP activation, induces PFT of vascular pericytes with conversion in fibroblasts followed by migration away from the vasculature to matrix stiffness. More details regarding plasticity can be found in the Supplement (Supplement, part 10, Cell transition plasticity).
CAFs and metastasis-associated fibroblasts (MAFs) CAF/MAF differentiation in MMT plays a key role in peritoneal metastasis [279]. In mesothelial cells, MMT is an important source of MAFs and peritoneal metastasis in CRC [273]. Based on their diverse cellular origins MAFs have been classified into myofibroblastic MAF populations; growth factor and inflammatory gene-expressing MAF populations; and portal fibroblast/mesothelial MAF populations, according to a modified CAF single-cell RNA sequencing signature [280]. These findings explain why MAFs show dynamically changing transcriptome profiles during metastasis [281]. MAFs drive not only carcinogenesis but also extracellular remodeling in metastatic epithelial cancers [282, 283]. In vitro studies have indicated that CRC-induced peritoneal metastasis originates from MAFs with high levels of IGFBP2 inducing CD38 plus immunosuppression by extracellular adenosine [284]. Application of these findings to available clinical data has revealed that MAFs are associated with poor prognosis. MAFs proliferate and migrate more than metastatic epithelial breast cancer cells [285]. In liver metastasis, these processes are promoted by PDGF, hyaluronic acid, and HGF [286]. In contrast to CAFs, MAFs express CD38 [287] and secrete higher levels of insulin-like growth factor-binding protein 2 (IGFBP2). Furthermore, CXCL12 and MAFs enhance breast cancer growth and metastasis more than CAFs alone in vivo [288]. IGFBP2 is immunosuppressive and stimulates IGF1R signaling. Therefore, colony formation of cancer cells is increased by MAFs, together with migration and invasion, and is more potent than that indiced by CAFs alone. MAFs are not only triggered by CAFs but also are induced by EVs [289–291], resident fibroblasts at distant sites [293–297], omental fibroblasts [298], HSCs [299], mesenchymal stem cells (MSCs) [300, 301], mesothelial cells (MCs) [273, 302], endothelial cells [259, 303], stromal cells derived from primary tumors [295], bone-marrow derived MSCs (BM-MSCs) [304], adipocytes [305], and even pericytes [306]. CAFs have very high plasticity [307]. Although CXCL10, CCL8, and metalloproteinase 3 (MMP-3) are secreted predominantly by CAFs, MAFs secrete higher levels of IGFBP2, CXCL12, and CXCL2 [287]. More detailed information regarding CAF-MAF signaling and crosstalk can be found in the Supplement (Supplement, part 11, CAFs and metastasis-associated fibroblasts). One undervalued pro-metastatic and pro-tumorigenic approach is direct or indirect immune escape. In this regard, another example immune evasion escape strategy in cancer involves sialic shielding. Pancreatic cancer cells cloak themselves with sialic acid: integrin α3β1, which is composed of ITGA3 and ITGB1 subunits, bind on the receptor Siglec-10, followed by inhibitory signaling to TAMs [308]. Siglec-10 was identified, cloned, and characterized in 2001 [309, 310]. Siglec-10 and cluster of differentiation 24 (CD24) suppress the immune response after tissue damage [311]. The cell adhesion molecule CD24 is expressed on epithelial cancers, such as those of the breast, lung, pancreas, prostate, colorectum, kidney, and ovary [312, 313]. This type of immune escape through pro-tumorigenic macrophage (M2) suppression has been demonstrated with CD24 and Siglec10 crosstalk in breast and ovarian cancers [314]. Siglec-10 is associated with poor survival in HCC [315], and inhibiting Siglec-10 in HCC M2s results in immune escape [315, 316]. Such shielding of tumors has also been shown in leukemia cells [317]. On the basis of careful observations by biochemists and pathologists dating back to Virchow, many beliefs were declared as “hallmarks” or “breakthroughs” in cancer and metastasis. Apoptosis provides an example. Cancers have long been known to have elevated frequencies of apoptosis [318]. Cancer cells are short (not long)-lived as also observed decades ago by the surgeon Alexis Carrel (1873-–1944) and the pathologist Albert Fischer (1891–1956) [319, 320]. Therefore, the notion that resistance or avoidance of apoptosis is a hallmark of cancer is incorrect. The consequence of unstoppable cell proliferation is, in fact, an increased rate of apoptosis. For creation of tissue, cells must exit proliferation by entering G0 stage, but as more cells proliferate, and more cells die, the G0 cell ratio decreases, thus resulting in decreased differentiation and structure, and increased cancer proliferation rates, necrosis rates, and apoptotic index values as cancer cells increasingly die [321]. Therefore, Bcl-2 is not elevated in aggressive breast or colon cancers. Interestingly, in this regard, Francis Peyton Rous (1879–1970), who was awarded the Nobel Prize in 1966 for the discovery of Rous sarcoma virus in 1911 [322], rejected the belief that cancer could be a genetic disease [323, 324]. Consequently, others wrongly judged Rous to be stubborn [325]. In our view, Rous was not stubbornly ignoring facts; instead, the authors who judged him a century later refused to accept the reality that he had observed.
Essential predisposing conditions for metastases The formation of three metastatic niches, as discussed above, creates essential conditions for metastases. Each metastatic niche is formed through multiple factors, including chronic inflammation, remodeling, signaling, and crosstalk, as summarized below.
Metastatic niche 1 (MN-1) (FIGURE 1) Metastatic niche 1 (MN-1) develops from the pre-metastatic niches (PMNs) [39]. Cancer satellites extravasate after traveling and nidate at the capillary bed after transendothelial migration, when de-coating into constituent parts occurs with EMT, MET, MMP, recruitment of immunocompetent cells, senescence, and dormancy (Fig. 1).
Metastatic niche 2 (MN-2) (FIGURE 2) The transformation of metastatic niche 1 (MN-1) to metastatic niche 2 (MN-2) is induced by chronic inflammation, fibrosis, and remodeling (via lysyl oxidase). Pro-tumorigenic TACs, EVs, and chemokine ligand 2 (CLL2) induce immune suppression. Granulin, periostin, fibronectin, CXCL12, and K19 increase motility, and the migration of non-dormant CAFs and cancer cells (Fig. 2). Further recruitment of fibroblasts induces LOX, which is followed by collagen crosslinking through oxidation of lysine residues, increased stiffness, and decreased elasticity. LOX also induces FAK, p130(cas)/crk/DOCK180 formation, which in turn increases lamellipodia/motility. Previously recruited macrophages, platelets, and neutrophils lead to pro-tumorigenic TACs with TAMs (M2), TAPs, and TANs. CAFs secrete EVs and CCL2, which in turn lead to the transformation of monocytes into MSCs, as well as immune suppression via decreased CD8+ T-cells and increased CD4+ TReg cells. These changes in the microenvironment result in consistent increases in granulosin, periostin, CXCL12, fibronectin, and K19, thus further increasing motility and promoting cancer cell migration.
Metastatic niche 3 (MN-3) (FIGURE 3) Metastastic niche 3 (MN-3) finally develops in which cell plasticity increases, active elongated cancer cells are present, and the newly developed MAFs re-trigger dormancy. Current anticancer therapies such as surgery trauma, chemotherapy, radiotherapy, immune therapy, and ongoing inflammation induce dormancy awakening, gelsolin with atypical EMT to spherical non-active cancer cells, followed by dormancy once more with increased heterogeneity, leading to metastatic cancer satellites and to immune suppression (Fig. 3). Therefore, the predisposing conditions for metastases are fulfilled: (1) metastatic cancer cells and (2) MAFs migrate along gradients; (3) metastatic cancer cells surrounded by MAFs are shielded from the immune system and consequently migrate to distant sites; (4) CXCL12 and K19 coat metastatic cancer cells; (5) platelets surround metastatic cancer cells, and (6) MAFs enable effective metastasis; (7) NETs shield metastatic cancer cells, and (8) MAFs, in parallel with ongoing dormancy and re-awakening, provide the basis for later metastasis. Distant metastatic niches create a high degree of heterogeneity of CAFs, MAFs, dormant and awakened cancer cells that progress to either metastatic cancer satellites that meet the predisposing conditions for metastases, or lead to dormancy.
Summary
It is an integral part of human existence and science to try to find answers to open questions, and if these be extremely complex, to simplify . Here, the human mind is affected by a bias which prefers simple explanations to complex phenomena although many variables might better explain real world outcomes or events. Such biases distort reasoning when it comes to the complexities of metastases [326]. Theories of cancer and metastasis development, including the somatic mutation theory, the clonal evolution theory, the epigenetic theory, the immunologic theory, the Warburg paradigm, and the stem cell cancer theory, have each essentially been simplifications, although the perspectives gained from immunology and stem cell science are important and complex. We are aware that some in retrospect may state that we combined parts of such into Epistemology of the origin of cancer. Here, we assume not defending against such arguments . To us, providing a logical explanation for creating progress with primary cancer patient care as our goal was, and remains the primary focus, although not all questions had been answered, and new questions always come up in answering others. Our intent is not be to question the origin of hereditary or the 5% of cancers caused by mutation, or the success of the 10% of cancers, such as leukemias, lymphomas, myelomas, or melanomas. During the past few decades, knowledge from genetics/epigenetics/proteomics has substantially increased. Here, disciplines have increased our knowledge in signaling and crosstalk in cancer biology. What is necessary in cancer science is to explain why and how some 80% of epithelial cancers occur, as genetic, epigenetic, and proteomic analyses alone may be insufficient to explain their initiation, progression, and metastatic behavior without considering epithelial physiology, tissue architecture, and stromal interactions. Although many genes, epigenetic changes, proteins, and molecular pathways contribute to epithelial carcinogenesis, no single molecular event or universal set of molecular events has been shown to account for the full initiation, progression, and metastatic behavior of human epithelial cancers. Cancer and metastasis are essentially states of disruption of homeostasis with complex signaling and crosstalk. Addressing fundamental questions in carcinogenesis and metastasis provide a much more nuanced, yet logical explanation: epithelial cancer develops predominantly from (1) a pathogenic stimulus, (2) chronic inflammation, (3) fibrosis and remodeling, and a (4) PCN detail [16, 17]. As mentioned, this sequence of events was later independently demonstrated in an animal model [36]. (5) Chronic stress escape strategy (CSES) lead by EMT into CAFs. (6) Finally, CAFs, aided by MET, can differentiate into cancer cells, and epithelial markers facilitate their integration into the epithelium [37]. The credit of the concept, that fibroblasts are the initial precursors of cancer, goe back to Virchow 168 years ago [38]. Metastasis is a complex phenomenon. Ongoing disruption of homeostasis with signaling and crosstalk transforms the PCN to PMNs during carcinogenesis [39]. PMN-1 involves p130 (cas)/crk/DOCK180 formation and consequent formation of lamellipodia, which enable cell mobility. As CAFs release fibronectin, CXCL12, and K19, the PMN-1 is transformed into PMN-2, with the transformation of anti-tumorigenic into pro-tumorigenic TAPs, TAMs (M2), and TANs. Persistent signaling and crosstalk induce immune evasion, and the transition of PMN-2 into PMN-3. Eight cancer satellites serve as prerequisites for intravasation/traveling: (1) cancer cells and (2) CAFs migrate along the CXCL12 and fibronectin gradient; (3) cancer cells surrounded by CAFs are shielded from the immune system and consequently travel; (4) CXCL12 and K19 coat cancer cells; (5) platelets surround cancer cells and (6) CAFs; and (7) NETs shield cancer cells and (8) CAFs [39]. Therefore, the PMNs create the conditions for the extravasation of heterogeneous cells and components. After transendothelial migration, cancer satellites travel and, if not eliminated by the immune system, extravasate after arriving at vascular endothelial capillaries. Furthermore, when cells grow, transendothelial migration occurs synchronously with local micro-metastasis. At this stage, metastatic colonization is observable under the microscope. The dissemination of metastatic cancer satellites is the major starting point for metastasis. Metastatic cancer cell heterogeneity is markedly increased by plasticity, as well as by the ongoing interplay between dormancy and reawakening; the transformation of CAFs to MAFs; and elongated active, and spherical inactive cancer cells. The metastatic niches are important basic steps in metastatic cell dormancy with secondary cancers with time delays. This metastatic dormancy can be seen in accordance with CD154/D40 signaling and crosstalk, CD40L transgene resulting in CD40 overexpression, initiating the SASP in non-small-cell lung cancer [98]. In brief, the high heterogeneity in cancer with its PMNs [39], and metastasis with its more complex metastatic niches (MNs), substantially increases heterogeneity. As discussed, this complexity might be a major reason why unidirectional views remain favored paradigms for cancer and metastasis despite incompletely explaining the disease. The vast heterogeneity may even limit universal rules, although in summary, the primary focus was, and remains, to develop a logical framework for advancing cancer research with improved patient care as the central goal. We acknowledge that our work does not answer all questions and new questions will arise. Opening new book chapters reveal limitations on various levels, e.g in regard to translational model(s) and/or an indirect proof as a logical method (if the opposite is impossible or leads to a contradiction). The analyzed knowledge pieced together here was provided by independent hundreds of separate studies in animal and human cancers. Despite the independent demonstration of the sequence of events of Part I [36], so far no single experiment has validated the full framework. Clinically we do not have a method to determine in which niche a cancer patient is in, or have verificated potential additional blood or imaging markers in this regard. We also do not have measurable criteria or markers to identify or differentiate boundaries between dormancy, quiescence, and senescence in clinical samples. Exemplary, real-time dynamics in cell plasticity will be a challenge, as there can be intermediate states [327] and e.g., EMT cancer cells very rapidly can polarize toward a mesenchymal or epithelial phenotype [328]. Therefore, the vast heterogenous interplay of adaptable changes in protein, subcellular and cellular compartements can range from micro-to-milliseconds, minutes, days, weeks, months or even years, because nature is never constant as assumed in models but consistently dynamic to maintain homeostasis. Nevertheless, a brief overview of implications to minimize or prevent metastasis were provided earlier [39].
Conclusions
Epithelial cancer and distant metastasis involve highly heterogeneous cancer cells, CAFs, MAFs, cell components, signaling, and crosstalk, which remain incompletely understood in the context of how their interactions promote disease. Regarding primary cancer and metastasis, each cell type is clinically recognized through histopathology, immunohistochemistry, and molecular biology, but their collective effects remain to be parsed. The fourth unanswered question in cancer biology concerns the essential conditions that permit metastasis to develop, including the role of tumor and microenvironmental heterogeneity. The ongoing disruption of homeostasis lies at the core of development of metastatic niches (MNs) and their sequential progression to metastases as illustrated in our Graphical Highlights and figures (Figs. 1 to 3).
The essential predisposing conditions for metastases are as follows: Cancer satellites from PMNs result in the first metastatic niche (MN-1), with nidation at the capillary bed, transendothelial migration, de-coating into constituent parts, recruitment of immunocompetent cells, cell transition via EMT and MET, and dormancy (Fig. 1). Metastastic niche 1 (MN-1) transforms into metastastic niche 2 (MN-2) with chronic inflammation, fibrosis and remodeling, TAMs, EVs, CLL2 with immune suppression, and increased motility of non-dormant cancer cells and CAFs along with granulin, periostin, fibronectin, CXCL12, and K19. (Fig. 2). Metastastic niche 3 (MN-3) is characterized by elevated heterogeneity due to plasticity, with development of MAFs, elongated active and spherical non-active cancer cells, and finally to heterogeneous metastatic cancer satellites with Trojan horses that facilitate immune evasion, and the interplay of dormancy and reawakening, alongside specific sequences metastasize alone or in combination at the distant site (Fig. 3): (1) metastatic cancer cells and (2) MAFs migrate along gradients; (3) metastatic cancer cells surrounded by MAFs are shielded from the immune system and consequently migrate to distant sites; (4) CXCL12 and K19 coat metastatic cancer cells; (5) platelets surround metastatic cancer cells and (6) MAFs, thus enabling effective metastasis; and (7) NETs shield metastatic cancer cells and (8) MAFs, in parallel with ongoing dormancy and re-awakening, thus forming the basis for later metastases. Plasticity is not only a major reason for the high heterogeneity in cancer and metastasis early during carcinogenesis [39] but also has been demonstrated to emerge early during colorectal carcinogenesis and to be driven by fibroblasts, which direct the timing and the location of plasticity [329]. Furthermore, inhibition of transcription factor EB (TFEB) through binding to 14-3-3σ, a frequently inactive regulatory p53 tumor suppression protein in CRC, promotes MET and inhibits autophagy in CRC [330]. The H1R antagonist, cetirizine, suppresses CAF-mediated remodeling and plasticity through signaling and crosstalk via COL1A1, COL3A1, fibronectin, FN1, heparan sulfate proteoglycan 2 (HSPG2), and TGFb in breast cancer cells (4T1). These findings suggest that the microenvironment regulates cell transition and metastasis [331]. CAF activation can be disrupted by photodynamic therapy, via CAF- mediated IGF1-IGF1R signaling and upregulation of RGS2, which in turn suppresses cancer progression [332]. Anticancer therapy, cancer surgery, and inflammation can favor the creation of MNs, thus explaining the frequent clinical observation of early appearing or later increased metastasis after a primary tumor undergoes surgery. This aspect also explains the paradoxical findings of a mixed response to anticancer therapy, and observations of metastasis with various mixed aggressive epithelial carcinomas with various rates and compositions of mesenchymal cell types and with various phenotypes. Furthermore, it sheds light on why highly metastatic epithelial cancers frequently show a highly mixed response in terms of the primary tumor versus metastasis in the lymph nodes or liver, lung, bones, or other locations. The observations in metastatic cancers explain why Isaiah Josh Fidler (1936–2020), a grandfather of metastasis research, wisely concluded in 1979 that “the search for a property uniform to all metastatic cells may be unproductive” [333], although “multiple cancer drugs have received “breakthrough therapy” designation, none to date have been safer, more effective, or more novel than drugs without this designation” [2]. Metastasis rates in epithelial cancers have not changed during the past century [2]. Perhaps it is time to recall the words of Henry Louis Mencken (1880-1956), who published an article in 1917, and later a book [334, 335], stating: "Explanations exist; they have existed for all time; there is always a well-known solution to every human problem—neat, plausible, and wrong." Simplification of models of metastasis to better understand complex cascades in a highly controlled environment has yielded only limited insights in vitro, because the in vivo situation is much more complex. In this regard, in 1877, Theodor Billroth (1829–1894) stated: “Nature is rarely dealt with in a simple ‘either-or’ way; it's all much more complicated than it first appears, and that we would like” [English translation] [336]. However, biology in humans is dynamic and never in a steady state. Various cancer models are excellent for identifying biochemical signaling or crosstalk. Otherwise, such have their limitations, e.g., having different aggressiveness, not capturing the interplay of dormancy and its awakening, individual steps during metastasis, or the complexity of participating cells, compartments, or the immune system, or their heterogeneity. Oversimplification might be a major reason for the currently limited understanding of howto better approach and treat epithelial cancers and their metastases. As illustrated in our Graphical Highlights and the figures, the suggested sequence of steps leading to distant metastatic niches is summarized in Figs. 1–3, illustrating the genesis of high heterogeneity of CAFs, MAFs, dormant and awakened cancer cells which progress to metastatic cancer satellites, and these aid in fulfilling the predisposing conditions for metastases. Additionally, this heterogeneity is deciphered to explain why no single anticancer targeted therapy approach can be successful, and a much more nuanced and differentiated anticancer therapy approach is necessary, as partially described earlier [39].
Abbreviations
12-HETE 12-hydroxyeicosatetraenoic acid; 20-HETE 20-hydroxyeicosatetraenoic acid, (5Z,8Z,11Z,14Z)-20-hydroxyicosa-5, 8,11, 14-tetraenoic acid; αIIbβ3 platelet glycoprotein IIb/IIIa integrin; αSMA alpha smooth muscle actin; ActA Activin A; Adgre1 adhesion G protein-coupled receptor E1, F4/80; AKT protein kinase B; ALOX arachidonate lipoxygenase; ANGPTL1 angiopoietin-related protein 1; AP-1 activator protein 1; Arg1 arginase 1; bFGF basic fibroblast growth factor; BLBCs basal-like breast cancers; BMP-1 bone morphogenetic protein-1; CAF cancer-associated fibroblast; CCC cholangiocellular carcinoma; CCL2 chemokine (C-C motif) ligand 2; CD31 cluster of differentiation 31, platelet endothelial cell adhesion molecule, PECAM-1; CD40 cluster of differentiation 40; CD44 cluster of differentiation 44 cell-surface glycoprotein; CD47 cluster of differentiation 47, integrin associated protein (IAP); CD62E ligand for E-selectin; CD99 cluster of differentiation 99, MIC2, single-chain type-1 glycoprotein; CD99L2 transmembrane protein CD99 molecule like 2; CD154 cluster of differentiation 154; CD202B cluster of differentiation 202B, TEK tyrosine kinase (TIE2); CD206 cluster of differentiation 206, mannose receptor; CD340 cluster of differentiation 340, human epidermal growth factor receptor 2 (HER2/neu), erythroblastic oncogene B receptor tyrosine-protein kinase erbB-2 (ErbB2); cdk cyclin-dependent kinase; C/EBPβ CCAAT/enhancer-binding protein beta; ck2 cyclin-dependent kinase 2; CLEC-2 C-type lectin-like receptor 2; Clec4f C-type lectin domain family 4 member F; CLL2 chemokine ligand 2; COL1A1 collagen type I; COL11A1 collagen type XI alpha I chain; COL13 collagen-13; Cox cyclooxygenase; Cox-1 cyclooxygenase 1; Cox-2 cyclooxygenase 2; CRC colorectal cancer; crk adapter molecule (p38); CSES chronic stress escape strategy; CSF1R colony stimulating factor 1 receptor; CTC circulating tumor cells; CUP cancer of unknown primary; CYP* cytochrome P450 isoforms; CXC CC chemokine receptors; CXCR4 C-X-C motif of chemokine receptor 4; CXCL1 C-X-C motif chemokine 1, GRO-α; CXCL4 C-X-C motif chemokine4, platelet factor 4 (PF4); CXCL5 C-X-C motif chemokine 5, Epithelial-derived neutrophil-activating protein 78, ENA-78; CXCL7 C-X-C motif chemokine 7, platelet basic protein; CXCL10 C-X-C motif chemokine ligand 10; CXCL12 C-X-C motif chemokine 12, stromal cell-derived factor 1 (SDF-1), pre-B cell growth-stimulating factor (PBSF); DCs antigen presenting dendritic cells; DmiR dormancy-associated miR; DOCK180 dedicator of cytokinesis; E2F4/5 cytoplasmic complex of Smad3, retinoblastoma-like protein 1 (P107, RBL1), E2F4/5 and D-prostanoid (DP1); EBV Epstein-Barr virus; EBF1 early B-cell factor 1; E-cadherin CAM 120/80 or epithelial cadherin, cadherin-1, epithelial cadherin; EMT epithelial-mesenchymal transition; EMP1 epithelial membrane protein 1; ENA-78 Epithelial-derived neutrophil-activating protein 78, C-X-C motif chemokine 5; EndMT endothelial-mesenchymal transition; ENO2 enolase 2; EpCAM epithelial cell adhesion molecule, cluster of differentiation 326, CD326); ER estrogen receptor; ErbB2 erythroblastic oncogene B receptor tyrosine-protein kinase erbB-2, human epidermal growth factor receptor 2 (HER2/neu), cluster of differentiation 340 (CD340); ERK extracellular signal-regulated kinase; ESAM endothelial cell adhesion molecule; ESCC esophageal squamous cell carcinoma; ET-B endothelin receptor type B (ETB1); EV extravesicular vesicle; F4/80 adhesion G protein-coupled receptor E1, Adgre1; FADS2 fatty acid desaturase 2, ω-6-desaturase (D6D); FAK focal adhesion kinase; FAP fibroblast activation protein; Fas ligand (FasL); FGF21 fibroblast growth factor 21; FGFR4 fibroblast growth factor (FGF) receptor 4; FOXO3 Forkhead box protein; FSP1 fibroblast‐specific protein 1, S100 calcium binding protein A4, S100A4; GARP glycoprotein A repetitions predominant; gas6 growth arrest-specific protein 6; G-CSF granulocyte colony-stimulating factor; GhHSP24.7 Gossypium hirsutum (Gh) heat shock protein (HSP) with approximate molecular weight 24.7 in kilodaltons (kDa); GLB1 galactosidase beta 1; GPCR adhesion G protein-coupled receptor (high-mobility group box 1, HMGB1); GREM1 gremlin-1; GRO-α C-X-C motif chemokine 1, CXCL1; HB-EGF heparin-binding epidermal growth factor; HCC hepatocellular carcinoma; HER2/neu human epidermal growth factor receptor 2; HGF hepatocyte growth factor; HMGB1 adhesion G protein-coupled receptor (high-mobility group box 1, GPCR); HSCs hepatic stellate cells; Hsp47 heat shock protein 47; IAP integrin associated protein, cluster of differentiation 47 (CD47); IFNγ interferon gamma; IL-1β interleukin 1 beta; IL-4 interleukin 4; IL-6 interleukin 6; IL-8 interleukin 8 (CXCL8); IL-26 interleukin 26; IL-27 interleukin 27; IL-33 interleukin 33; ICAM-1 intercellular adhesion molecule 1, CD54; INHBA inhibin subunit beta A; IGFBP1 insulin-like growth factor binding protein 1; ITGA11 integrin α11β1; ITGB4 integrin β4; JAX Jackson laboratory; JNK c-Jun NH2-terminal kinase; KCs resident Kupffer cells, KC1 (CD45+ F4/80+ CD11bInt TIM-4+ ESAM-) and KC2 (CD45+ F4/80+ CD11bInt TIM-4+ ESAM+); K19 keratin 19; LMP1 latent membrane protein 1; LOX lysyl oxidase; LPS lipopolysaccharide; LSEC liver sinusoidal endothelial cell; LTBP1 latent transforming growth factor beta-binding protein 1; LTs leukotrienes; LYVE1 lymphatic vessel endothelial hyaluronan receptor 1; MAGE-A3/6 melanoma-associated antigens A3 and A6; MCs mesothelial cells; MDSCs myeloid-derived suppressor cells; MesoMT mesothelial-mesenchymal transition; MET mesenchymal-epithelial transition; MFAP5 microfibril associated protein 5; MHC major histocompatibility complex; MIIP migration and invasion inhibitory protein (IIP45); miR21 microRNA-21; MMP mesenchymal-mesenchymal plasticity; MMPs metalloproteinases; MMP-3 metalloproteinase 3; MMP-9 metalloproteinase 9; MMP-12 metalloproteinase12, macrophage elastase; MMT mesothelial-mesenchymal transition; MNs metastatic niches; MN-1 metastatic niche 1; MN-2 metastatic niche 2; MN-3 metastatic niche 3; MSCs mesenchymal stem cells; MSLN mesothelin, CAK1 antigen, pre-pro-megakaryocyte-potentiating factor; mTORC1 rapamycin complex 1; NASH non-alcoholic steatohepatitis; NCCCT normal cell to cancer cell transition; NETs neutrophil extracellular traps; NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells; NK natural killer T-cells; NPY neuropeptide Y; NPY1R neuropeptide Y (NPY) receptor Y1; NUPR1 nuclear stress protein nuclear protein 1, p8; NURF nucleosome remodeling factor; OL oral leukoplakia; OSCC oral squamous cell carcinoma; p16 cyclin-dependent kinase inhibitor 2A (p16, p16INK4a); p21 cyclin-dependent kinase inhibitor 1, alternatively p21Waf1; p38 MAPK mitogen-activated protein kinase; p53 tumor protein p53; p107 retinoblastoma-like protein 1, RBL1; p130(cas) breast cancer anti-estrogen resistance protein 1, BCAR1; p300 protein 300, p300-CBP coactivator family; PAI1 plasminogen activator inhibitor-1; PBSF pre-B cell growth-stimulating factor, C-X-C motif chemokine 12 (CXCL12), stromal cell-derived factor 1 (SDF-1); PCN precancerous niche; PDGF-β platelet-derived growth factor beta; PDPN podoplanin; PECAM-1 platelet endothelial cell adhesion molecule, CD31, Cluster of differentiation 31; PF4 platelet factor 4 (C-X-C motif chemokine4, CXCL4); PFT pericyte-fibroblast transition; PGD2 prostaglandin D2, (Z)-7-[(1R,2R,5S)-5-hydroxy-2-[(E,3S)-3-hydroxyoct-1-enyl]-3-oxocyclopentyl]hept-5-enoic acid; PGE2 prostaglandin E2, (Z)-7-[(1R,2R,3R)-3-hydroxy-2-[(E,3S)-3-hydroxyoct-1-enyl]-5-oxocyclopentyl]hept-5-enoic acid; PGF2 prostaglandin F2 alpha, (Z)-7-[(1R,2R,3R,5S)-3, 5-dihydroxy-2-[(E,3S)-3-hydroxyoct-1-enyl]cyclopentyl]hept-5-enoic acid; PKR-1 prokineticin receptor; PPG2 prostaglandin G2, (Z)-7-[(1S,4R,5R,6R)-5-[(E,3S)-3-hydroperoxyoct-1-enyl]-2, 3-dioxabicyclo [2.2.1]heptan-6-yl]hept-5-enoic acid; PGH2 prostaglandin H2, (Z)-7-[(1S,4R,5R,6R)-5-[(E,3S)-3-hydroxyoct-1-enyl]-2, 3-dioxabicyclo [2.2.1]heptan-6-yl]hept-5-enoic acid; PGI2 prostaglandin I2, prostacyclin I2, (5Z)-5-[(3aR,4R,5R,6aS)-5-hydroxy-4-[(E,3S)-3-hydroxyoct-1-enyl]-3, 3a,4, 5,6, 6a-hexahydrocyclopenta [b]furan-2-ylidene]pentanoic acid; PI3K phosphatidylinositide 3-kinase; PLT-Exos exosomes derived from platelets; PMN premetastatic niche; PMN-1 pre-metastatic niche 1; PMN-2 pre-metastatic niche 2; PMN-3 pre-metastatic niche 3; POSTN periostin, former osteoblast-specific factor 2; PPARγ peroxisome proliferator-activated receptor γ; PRKCG protein kinase C gamma gene; PTPase protein tyrosine phosphatase; RANKL receptor activator of NF-κB ligand; RhoA Ras homolog family member A; RIGS radioimmune-guided surgery; ROS reactive oxygen species; S1P sphingosine-1-phosphate; S100A4 S100 calcium binding protein A4, fibroblast‐specific protein 1, FSP1; S100A8 calcium-binding protein A8; S100A9 calcium-binding protein A9 (S100A9), migration inhibitory factor-related protein 14 (MRP14); SAβ-gal senescence-associated β-galactosidase; SASP senescence-associated secretory phenotype; SCLC small cell lung cancer; SCRNA seq single-cell RNA sequencing; SDF-1 stromal cell-derived factor 1, C-X-C motif chemokine 12 (CXCL12), pre-B cell growth-stimulating factor (PBSF); sE-cad soluble CAM 120/80 (E-cadherin); SERPINE1 Serpin Family E Member 1; S-Gal beta-galactosidase-related protein; Shp2 Src-homology 2 domain-containing tyrosine phosphatase 2; SIRT1 mammalian sirtuin 1; Snail zinc finger protein SNAI1; SOX [sex-determining region Y (Sry) box-containing] transcription factor family; SP1 specificity protein 1; SPARC secreted protein acidic and rich in cysteine, osteonectin; SPP1 phosphoprotein 1; STAT3 transcription factor activator protein 3; TAC tumor-associated cell; TAGLN transgelin; TAM tumor-associated macrophage; TAN tumor-associated neutrophil; TAP tumor-associated platelet; TENs tumor-entrained neutrophils; TGFβ transforming growth factor beta; TGFβ2 secreted protein, transforming growth factor beta2; TH T-helper cells, with various subsets Th1, Th2, Th17 and suppressing CD4+ regulatory T-cells (TReg); TIE2 TEK tyrosine kinase, cluster of differentiation 202B (CD202B); Tim4 T-cell immunoglobulin and mucin domain containing 4; TLR9 Toll-like receptor 9; TMT1A thiol methyltransferase 1A; TNFa tumor necrosis factor alpha; TReg suppressing CD4+ regulatory T-cells; TSP1 thrombospondin-1; TXA2 thromboxane A2, (Z)-7-[(1S,2S,3R,5S)-3-[(E,3S)-3-hydroxyoct-1-enyl]-4, 6-dioxabicyclo [3.1.1]heptan-2-yl]hept-5-enoic acid; VCAM-1 vascular cell adhesion protein 1, cluster of differentiation 106 (CD106); VE-cadherin vascular endothelial-cadherin; VEGF vascular endothelial growth factor; VIRMA vir-like m6A methyltransferase-associated protein (KIAA1429); YAP yes-protein.
Acknowledgements
This paper is dedicated to the memory of a wonderful and gentle human, scientist and active member of the Theodor-Billroth-Academy®, Munich, Germany, and Sacramento, CA, USA, and its International Consortium of Research Excellence: Dr. Ray Perkins (1951-2026), New Liberty Proteomics Corporation, New Liberty, KY, USA, who passed away unexpectedly on Mar 14, 2026. We are thankful to the countless scientists, clinicians, and individuals of various disciplines and professions from Asia, Europe, North and South America, and Australia for personal exchanges during the past few decades. We acknowledge the intense and bias-free discussions, critical thinking, exchanges, and independent critical pro bono peer review provided to us by Prof. em. Dr. Masaharu Seno, Department of Cancer Stem Cell Engineering, Faculty of Interdisciplinary Science and Engineering in Health Systems, Okayama University, Okayama, Japan (ORCID: 0000-0001-8547-6259); Prof. em. Dr. Jose Florencio F Lapeña Jr., Otolaryngologist, World Association of Medical Editors, Manilla, the Philippines (ORCID: 0000-0002-5794-1878); Prof. Dr. Marjan Slak Rupnik, Physiologist & Pathophysiologist, Vienna, Austria (ORCID: 0000-0002-3744-4882); Prof. Dr. Volkmar Weissig, Department of Pharmaceutical Sciences, Midwestern University, College of Pharmacy, Glendale, AZ, USA (ORCID: 0000-0002-6466-2367); Prof. Dr. Oliver Hayden, Heinz-Nixdorf-Chair of Biomedical Electronics, TranslaTUM, Department of Electrical and Computer Engineering, TU Munich, Munich, Germany (ORCID: 0000-0002-2678-8663); Dr. Gudrun Schüler, Hematologist & Oncologist, Cottbus, Germany; Prof. Dr. Lúcio Lara Santos Surgical Oncology Department, Portuguese Institute of Oncology, Porto, Portugal (ORCID: 0000-0002-0521-5655); Dr. José M Correia da Costa, National Institute of Health Ricardo Jorge, Porto, Portugal (ORCID: 0000-0001-6591-4303). Each of these individuals has approved this acknowledgment. The creation of this article was supported by the Theodor-Billroth-Academy®, Munich, Germany, and Sacramento, CA, USA, its International Consortium of Research Excellence, and the Cancer Metastases Research Fund.
Disclosure Statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this manuscript. This manuscript contains original material that has been previously published and is appropriately cited. The opinions or assertions contained herein are those of the authors alone and are not to be construed as official or reflecting the views of the publisher or their employers. BB wrote the first MS draft. Both authors contributed to the content and approved the final version of the manuscript. The manuscript was created from our intellectual input only. No artificial intelligence software, e.g., ChatGPT or others, was used in the preparation of this manuscript.
References
- Roser M: “Twice as long — life expectancy around the world” Published online at OurWorldinData.org 2018. Retrieved from: https://archive.ourworldindata.org/20251125-173858/life-expectancy-globally.html [Online Resource] (archived on November 25, 2025; 21:17:23-43. https://doi.org/10.2147/JHL.S506767
- Brücher BLDM: The Erosion of Healthcare and Scientific Integrity: A Growing Concern. J Healthc Leadersh 2025;17:23-43. https://doi.org/10.2147/JHL.S506767
- Brenner H, Heisser T, Cardoso R, Hoffmeister M: Reduction in colorectal cancer incidence by screening endoscopy. Nat Rev Gastroenterol Hepatol. 2024;21(2):125–133. https://doi.org/10.1038/s41575-023-00847-3
- Botteri E, Holme Ø, Løberg M, Bretthauer M, Kalager M, Randel KR, Hoff G: Twenty-Three-Year Benefits of Sigmoidoscopy Screening for Colorectal Cancer: A Randomized Trial. Ann Intern Med 2026 May 12. https://doi.org/10.7326/ANNALS-25-05456
- Siegel RL, Wagle NS, Jemal A: Leading Cancer Deaths in People Younger Than 50 Years. JAMA. 2026 Jan 22. https://doi.org/10.1001/jama.2025.25467
- Lynch HT, Smyrk TC, Watson P, Lanspa SJ, Lynch JF, Lynch PM, Cavalieri RJ, Boland CR: Genetics, natural history, tumor spectrum, and pathology of hereditary nonpolyposis colorectal cancer: an updated review. Gastroenterology. 1993 May;104(5):1535-49. https://doi.org/10.1016/0016-5085(93)90368-m
- Liu B, Nicolaides NC, Markowitz S, Willson JK, Parsons RE, Jen J, Papadopolous N, Peltomäki P, de la Chapelle A, Hamilton SR, Kinzler KW, Vogelstein B: Mismatch repair gene defects in sporadic colorectal cancers with microsatellite instability. Nat Genet. 1995 ;9(1):48-55. https://doi.org/10.1038/ng0195-48
- Tomlinson IP, Novelli MR, Bodmer WF: The mutation rate and cancer. Proc Natl Acad Sci U S A. 1996 Dec 10;93(25):14800-3. https://doi.org/10.1073/pnas.93.25.14800
- Pisani P, Parkin DM, Muñoz N, Ferlay J: Cancer and infection: estimates of the attributable fraction in 1990. Cancer Epidemiol Biomarkers Prev. 1997 Jun;6(6):387-400 PMID: 9184771.
- Blattner WA: Human retroviruses: their role in cancer. Proc Assoc Am Physicians 1999;111(6):563-572. https://doi.org/10.1046/j.1525-1381.1999.99210.x
- Burt RW: Colon cancer screening. Gastroenterology 2000;119(3):837-853. https://doi.org/10.1053/gast.2000.16508
- Parkin DM: The global health burden of infection-associated cancers in the year 2002. Int J Cancer 2006;118(12):3030-3044. https://doi.org/10.1002/ijc.21731
- Vineis P: Misuse of genetic data in environmental epidemiology. Ann N Y Acad Sci 2006;1076:163-167. https://doi.org/10.1196/annals.1371.060
- Rustgi AK: The genetics of hereditary colon cancer. Genes Dev 2007;21(20):2525-38. https://doi.org/10.1101/gad.1593107
- Markowitz SD, Bertagnolli MM: Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med 2009;361(25):2449-60. https://doi.org/10.1056/NEJMra0804588
- Brücher BL, Jamall IS: Cell-cell communication in the tumor microenvironment, carcinogenesis, and anticancer treatment. Cell Physiol Biochem 2014;34(2):213-243. https://doi.org/10.1159/000362978
- Brücher BL, Jamall IS: Epistemology of the origin of cancer: a new paradigm. BMC Cancer 2014;14:331. https://doi.org/10.1186/1471-2407-14-331
- Chan JC: Hypotheses of cancer weakening and origin. J Cancer 2015;6(5):457-463. https://doi.org/10.7150/jca.10911
- Richman S: Deficient mismatch repair: Read all about it (Review). Int J Oncol 2015;47(4):1189-1202 https://doi.org/10.3892/ijo.2015.3119
- Brücher BL, Li Y, Schnabel P, Daumer M, Wallace TJ, Kube R, Zilberstein B, Steele S, Voskuil JL, Jamall IS: Genomics, microRNA, epigenetics, and proteomics for future diagnosis, treatment and monitoring response in upper GI cancers. Clin Transl Med 2016;5(1):13. https://doi.org/10.1186/s40169-016-0093-6
- Brücher BL, Jamall IS. Somatic Mutation Theory - Why it's Wrong for Most Cancers. Cell Physiol Biochem 2016;38(5):1663-1680. https://doi.org/10.1159/000443106
- Rappaport SM: Genetic Factors Are Not the Major Causes of Chronic Diseases. PLoS One 2016;11(4):e0154387. https://doi.org/10.1371/journal.pone.0154387
- Wu S, Powers S, Zhu W, Hannun YA: Substantial contribution of extrinsic risk factors to cancer development. Nature 2016;529(7584):43-47. https://doi.org/10.1038/nature16166
- Adjiri A: DNA Mutations May Not Be the Cause of Cancer. Oncol Ther 2017;5(1):85-101. https://doi.org/10.1007/s40487-017-0047-1
- Kanth P, Grimmett J, Champine M, Burt R, Samadder NJ: Hereditary Colorectal Polyposis and Cancer Syndromes: A Primer on Diagnosis and Management. Am J Gastroenterol 2017;112(10):1509-1525. https://doi.org/10.1038/ajg.2017.212
- Jonsson P, Bandlamudi C, Cheng ML, Srinivasan P, Chavan SS, Friedman ND, Rosen EY, Richards AL, Bouvier N, Selcuklu SD, Bielski CM, Abida W, Mandelker D, Birsoy O, Zhang L, Zehir A, Donoghue MTA, Baselga J, Offit K, Scher HI, O'Reilly EM, Stadler ZK, Schultz N, Socci ND, Viale A, Ladanyi M, Robson ME, Hyman DM, Berger MF, Solit DB, Taylor BS: Tumour lineage shapes BRCA-mediated phenotypes. Nature 2019;571(7766):576-579. https://doi.org/10.1038/s41586-019-1382-1
- de Martel C, Georges D, Bray F, Ferlay J, Clifford GM: Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob Health 2020;8(2):e180-e190. https://doi.org/10.1016/S2214-109X(19)30488-7
- Yang Z, Pajuste FD, Zguro K, Cheng Y, Kurant DE, Eoli A, Wanner J, Jermy B, Rämö J; FinnGen; Kanoni S, van Heel DA; Genes & Health Research Team; Hayward C, Marioni RE, McCartney DL, Renieri A, Furini S; INTERVENE consortium; Mägi R, Gusev A, Drineas P, Paschou P, Heyne H, Ripatti S, Mars N, Ganna A: Limited overlap between genetic effects on disease susceptibility and disease survival. Nat Genet 2025;57(10):2418-2426. https://doi.org/10.1038/s41588-025-02342-8
- Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K: Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 2000;343(2):78-85. https://doi.org/10.1056/NEJM200007133430201
- Brücher BLDM, Jamall IS: Chronic inflammation evoked by pathogenic stimulus during carcinogenesis. 4open 2019;2(8):1-22. https://doi.org/10.1051/fopen/2018006
- Brücher BL, Jamall IS: Precancerous niche (PCN), a product of fibrosis with remodeling by incessant chronic inflammation. 4open 2019;2(11):1-21. https://doi.org/10.1051/fopen/2018009
- Brücher BLDM, Lang F, Jamall IS: NF-κB signaling and crosstalk during carcinogenesis. 4open 2019;2(13):1-35. https://doi.org/10.1051/fopen/2019010
- Brücher BL, Jamall IS: Eicosanoids in carcinogenesis. 4open 2019;2(9):1-34. https://doi.org/10.1051/fopen/2018008
- Brücher BLDM, Jamall IS: Synopsis: Special Issue on "Disruption of signaling homeostasis induced crosstalk in the carcinogenesis paradigm Epistemology of the origin of cancer". 4open 2019;2(28):1-30. https://doi.org/10.1051/fopen/2019023
- Brücher BLDM, Daumer M, Jamall IS: Physics Essentials Enable Deeper Understanding in Signaling and Crosstalk of the Carcinogenesis Paradigm "Epistemology of the Origin of Cancer". Cell Physiol Biochem 2022;56(5):546-572. https://doi.org/10.33594/000000575
- Basree MM, Shinde N, Koivisto C, Cuitino M, Kladney R, Zhang J, Stephens J, Palettas M, Zhang A, Kim HK, Acero-Bedoya S, Trimboli A, Stover DG, Ludwig T, Ganju R, Weng D, Shields P, Freudenheim J, Leone GW, Sizemore GM, Majumder S, Ramaswamy B: Abrupt involution induces inflammation, estrogenic signaling, and hyperplasia linking lack of breastfeeding with increased risk of breast cancer. Breast Cancer Res 2019;21(1):80. https://doi.org/10.1186/s13058-019-1163-7
- Brücher BLDM, Jamall IS: Epistemology of the Origin of Cancer II: Fibroblasts Are the First Cells to Undergo Neoplastic Transformation. Cell Physiol Biochem 2023;57(6):512-537. https://doi.org/10.33594/000000672
- Virchow R: Die Cellularpathologie in Ihrer Begründung auf Physiologische und Pathologische Gewebelehre. Hirschwald August Verlag, Berlin. Page 410,1858.
- Brücher BLDM, Jamall IS: Epistemology of the Origin of Cancer III: Fundamentals of How Metastasis Arises. Cell Physiol Biochem 2025;59(6):753-799. https://doi.org/10.33594/000000826
- Recamier JC: Recherches sur le traitment du cancer, par la compression méthodique simple ou combinée, sur l_histoire générale de la méme maladie. Chez Gabon, Libraire-Editeur, Paris. 1829.
- Fuchs E: Das Sarkom des Uvealtractus. Habilitationsschrift, Wilhelm Braunmüller. Wien und Graefe's Arch Ophthalmol, XII, Wien. 1882.
- Paget S: The distribution of secondary growth in cancer of the breast. Lancet 1899;1:571–573. https://doi.org/10.1016/S0140-6736(00)49915-0
- Fidler JJ. Metastasis: Quantitative analysis of distribution and fate of tumor embolilabeled with 125 I-5-iodo-2′-deoxyuridine. J Natl Cancer Inst 1970;45(4):773–782. PMID: 5513503.
- Langley RR, Fidler IJ: The seed and soil hypothesis revisited-the role of tumor-stroma interactions in metastasis to different organs. Int J Cancer 2011;128(11):2527-35. https://doi.org/10.1002/ijc.26031
- Ashworth TR: A case of cancer in which cells similar to those of the tumour were seen in the blood after death. Med J Aust 1869;14:146–147.
- Goldmann EE: Anatomische Untersuchungen über die Verbreitungswege bösartiger Geschwülste. Beitr z klin Chir 1897;18:595.
- Zhe X, Cher ML, Bonfil RD: Circulating tumor cells: finding the needle in the haystack. Am J Cancer Res 2011;1(6):740–751. PMID: 22016824.
- Jones DS, Wallace AC, Fraser EE: Sequence of events in experimental metastases of Walker 256 tumor: light, immunofluorescent, and electron microscopic observations. J Natl Cancer Inst 1971;46(3):493-504. PMID: 4100699.
- Nieswandt B, Hafner M, Echtenacher B, Männel DN: Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res 1999;59(6):1295-1300. PMID: 1009656.2.
- Gasic GJ, Gasic TB, Stewart CC: Antimetastatic effects associated with platelet reduction. Proc Natl Acad Sci USA 1968;61(1):46-52. https://doi.org/10.1073/pnas.61.1.46
- Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR: Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood 2004;104(2):397-401. https://doi.org/10.1182/blood-2004-02-0434
- Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z: Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet 2012;379(9826):1591-1601. https://doi.org/10.1016/S0140-6736(12)60209-8
- Skolnik G, Alpsten M, Ivarsson L: Studies on mechanisms involved in metastasis formation from circulating tumor cells. Factors influencing tumor cell lodgement during normal and post-traumatic conditions. J Cancer Res Clin Oncol 1980;97(3):249-256. https://doi.org/10.1007/BF00405776
- Yeini E, Satchi-Fainaro R: The role of P-selectin in cancer-associated thrombosis and beyond. Thromb Res 2022;213 Suppl 1:S22-S28. https://doi.org/10.1016/j.thromres.2021.12.027
- Shaul ME, Eyal O, Guglietta S, Aloni P, Zlotnik A, Forkosh E, Levy L, Weber LM, Levin Y, Pomerantz A, Nechushtan H, Eruslanov E, Singhal S, Robinson MD, Krieg C, Fridlender ZG: Circulating neutrophil subsets in advanced lung cancer patients exhibit unique immune signature and relate to prognosis. FASEB J 2020;34(3):4204-4218. https://doi.org/10.1096/fj.201902467R
- Wang Z, Moresco P, Yan R, Li J, Gao Y, Biasci D, Yao M, Pearson J, Hechtman JF, Janowitz T, Zaidi RM, Weiss MJ, Fearon DT: Carcinomas assemble a filamentous CXCL12-keratin-19 coating that suppresses T cell mediated immune attack. Proc Natl Acad Sci USA 2022;119(4):e2119463119. https://doi.org/10.1073/pnas.2119463119
- Yan R, Moresco P, Gegenhuber B, Fearon DT: T cell-mediated development of stromal fibroblasts with an immune-enhancing chemokine profile. Cancer Immunol Res 2023;May 22:CIR-22-0593. https://doi.org/10.1158/2326-6066.CIR-22-0593
- Ito S, Nakanishi H, Ikehara Y, Kato T, Kasai Y, Ito K, Akiyama S, Nakao A, Tatematsu M: Real-time observation of micrometastasis formation in the living mouse liver using a green fluorescent protein gene-tagged rat tongue carcinoma cell line. Int J Cancer 2001;93(2):212-217. https://doi.org/10.1002/ijc.1318
- Rosso R, Lucioni M: Normal and neoplastic cells of brown adipose tissue express the adhesion molecule CD31 Arch Pathol Lab Med 2006;130(4):480-482. https://doi.org/10.5858/2006-130-480-NANCOB
- Higashi N, Ishii H, Fujiwara T, Morimoto-Tomita M, Irimura T: Redistribution of fibroblasts and macrophages as micrometastases develop into established liver metastases. Clin Exp Metastasis 2002;19(7):631-638. https://doi.org/10.1023/a:1020946300690
- Fukumura D, Yuan F, Monsky WL, Chen Y, Jain RK: Effect of host microenvironment on the microcirculation of human colon adenocarcinoma. Am J Pathol 1997;151(3):679-688. PMID: 9284816.
- Craig LE, Spelman JP, Strandberg JD, Zink MC: Endothelial cells from diverse tissues exhibit differences in growth and morphology. Microvasc Res 1998;55(1):65-76. https://doi.org/10.1006/mvre.1997.2045
- Simon T, Bromberg JS. Regulation of the Immune System by Laminins. Trends Immunol 2017;38(11):858-871. https://doi.org/10.1016/j.it.2017.06.002
- Song J, Zhang X, Buscher K, Wang Y, Wang H, Di Russo J, Li L, Lütke-Enking S, Zarbock A, Stadtmann A, Striewski P, Wirth B, Kuzmanov I, Wiendl H, Schulte D, Vestweber D, Sorokin L: Endothelial Basement Membrane Laminin 511 Contributes to Endothelial Junctional tightness and Thereby Inhibits Leukocyte Transmigration. Cell Rep 2017;18(5):1256-1269. https://doi.org/10.1016/10.1016/j.celrep.2016.12.092
- Sender R, Fuchs S, Milo R: Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016 Aug 19;14(8):e1002533. https://doi.org/10.1371/journal.pbio.1002533
- Sender R, Milo R: The distribution of cellular turnover in the human body. Nat Med. 2021 ;27(1):45-48. https://doi.org/10.1038/s41591-020-01182-9
- Moore CH: On the Influence of Inadequate Operations on the Theory of Cancer, Med Chir Trans 1867;50:245-280. https://doi.org/10.1177/095952876705000114
- Willis RA: Pathology of Tumours. Butterworth and Co. Ltd., London. 1948.
- Hadfield G: The dormant cancer cell. Br Med J 1954;2(4888):607-610. https://doi.org/10.1136/bmj.2.4888.607
- Damen MPF, van Rheenen J, Scheele CLGJ: Targeting dormant tumor cells to prevent cancer recurrence. FEBS J 2021;288(21):6286-6303. https://doi.org/10.1111/febs.15626
- Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H, Hara E: Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol 2006;8(11):1291-1297. https://doi.org/10.1038/ncb1491
- Keilin D: The problem of anabiosis or latent life: history and current concept. Proc R Soc Lond B Biol Sci 1959;150(939):149-191. https://doi.org/10.1098/rspb.1959.0013
- O'Farrell PH: Quiescence: early evolutionary origins and universality do not imply uniformity. Philos Trans R Soc Lond B Biol Sci 2011;366(1584):3498-3507. https://doi.org/10.1098/rstb.2011.0079
- Considine MJ, Considine JA: On the language and physiology of dormancy and quiescence in plants. J Exp Bot 2016;67(11):3189-3203. https://doi.org/10.1093/jxb/erw138
- Gomis RR, Gawrzak S: Tumor cell dormancy. Mol Oncol 2017;11(1):62-78. https://doi.org/10.1016/j.molonc.2016.09.009.
- Park SY, Nam JS: The force awakens: metastatic dormant cancer cells. Exp Mol Med 2020;52(4):569-581. https://doi.org/10.1038/s12276-020-0423-z
- Phan TG, Croucher PI: The dormant cancer cell life cycle. Nat Rev Cancer 2020;20(7):398-411. https://doi.org/10.1038/s41568-020-0263-0
- Coller HA, Sang L, Roberts JM: A new description of cellular quiescence. PLoS Biol 2006;4(3):e83. https://doi.org/10.1371/journal.pbio.0040083
- Cheung TH, Rando TA: Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol 2013;14(6):329-340. https://doi.org/10.1038/nrm3591
- Lennon JT, Jones SE: Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev Microbiol 2011;9(2):119-30. https://doi.org/10.1038/nrmicro2504
- Webster KD, Lennon JT: Dormancy in the origin, evolution and persistence of life on Earth. Proc Biol Sci 2025;292(2038):20242035. https://doi.org/10.1098/rspb.2024.2035
- Mangelsdorf PC: The Inheritance of Dormancy and Premature Germination in Maize. Genetics 1930;15(5):462-94. https://doi.org/10.1093/genetics/15.5.462
- Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD: Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007;450(7171):903-907. https://doi.org/10.1038/nature06309.
- Teng MW, Vesely MD, Duret H, McLaughlin N, Towne JE, Schreiber RD, Smyth MJ: Opposing roles for IL-23 and IL-12 in maintaining occult cancer in an equilibrium state. Cancer Res 2012;72(16):3987-3996. https://doi.org/10.1158/0008-5472.CAN-12-1337
- Minot CS: Senescence and Rejuvenation. J Physiol 1891;12(2):97-192.113. https://doi.org/10.1113/jphysiol.1891.sp000369
- Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J: Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003;22(16):4212-4222. https://doi.org/10.1093/emboj/cdg417
- Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA, Mendoza-Parra MA, Kanashova T, Metzner M, Pardon K, Reimann M, Trumpp A, Dörken B, Zuber J, Gronemeyer H, Hummel M, Dittmar G, Lee S, Schmitt CA: Senescence-associated reprogramming promotes cancer stemness. Nature 2018;553(7686):96-100. https://doi.org/10.1038/nature25167
- Otsuki L, Brand AH: Cell cycle heterogeneity directs the timing of neural stem cell activation from quiescence. Science 2018;360(6384):99-102. https://doi.org/10.1126/science.aan8795
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peqacocke M, Campisi J: A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995;92(20):9363-9367. https://doi.org/10.1073/pnas.92.20.936
- Kurz DJ, Decary S, Hong Y, Erusalimsky JD: Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci 2000;113 (Pt 20):3613-3622. https://doi.org/10.1242/jcs.113.20.3613
- Uhrbom L, Nistér M, Westermark B: Induction of senescence in human malignant glioma cells by p16INK4A. Oncogene 1997;15(5):505-514. https://doi.org/10.1038/sj.onc.1201227.
- Nakabayashi K, Ogata T, Fujii M, Tahara H, Ide T, Wadhwa R, Kaul SC, Mitsui Y, Ayusawa D: Decrease in amplified telomeric sequences and induction of senescence markers by introduction of human chromosome 7 or its segments in SUSM-1 Exp Cell Res 1997;235(2):345-353. https://doi.org/10.1006/excr.1997.3678
- Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, Kleijer WJ, DiMaio D, Hwang ES: Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006;5(2):187-195. https://doi.org/10.1111/j.1474-9726.2006.00199.x
- Duca L, Blanchevoye C, Cantarelli B, Ghoneim C, Dedieu S, Delacoux F, Hornebeck W, Hinek A, Martiny L, Debelle L: The elastin receptor complex transduces signals through the catalytic activity of its Neu-1 subunit. J Biol Chem 2007;282(17):12484-12491. https://doi.org/10.1074/jbc.M609505200
- Overgaard M, Hansen PS, Overgaard J, Rose C, Andersson M, Bach F, Kjaer M, Gadeberg CC, Mouridsen HT, Jensen MB, Zedeler K: Postoperative radiotherapy in high-risk premenopausal women with breast cancer who receive adjuvant chemotherapy. Danish Breast Cancer Cooperative Group 82b Trial. N Engl J Med 1997;337(14):949-955. https://doi.org/10.1056/NEJM1997100233714
- Ragaz J, Jackson SM, Le N, Plenderleith IH, Spinelli JJ, Basco VE, Wilson KS, Knowling MA, Coppin CM, Paradis M, Coldman AJ, Olivotto IA: Adjuvant radiotherapy and chemotherapy in node-positive premenopausal women with breast cancer. N Engl J Med 1997;337(14):956-962. https://doi.org/10.1056/NEJM199710023371402
- Gamucci T, Vaccaro A, Ciancola F, Pizzuti L, Sperduti I, Moscetti L, Longo F, Fabbri MA, Giampaolo MA, Mentuccia L, Di Lauro L, Vici P: Recurrence risk in small, node-negative, early breast cancer: a multicenter retrospective analysis. J Cancer Res Clin Oncol 2013;139(5):853-860. https://doi.org/10.1007/s00432-013-1388-2
- Xu W, Li Y, Yuan WW, Yin Y, Song WW, Wang Y, Huang QQ, Zhao WH, Wu JQ: Membrane-Bound CD40L Promotes Senescence and Initiates Senescence-Associated Secretory Phenotype via NF-κB Activation in Lung Adenocarcinoma. Cell Physiol Biochem 2018;48(4):1793-1803. https://doi.org/10.1159/000492352
- Pelosi G, Pasini F, Ottensmeier C, Pavanel F, Bresaola E, Bonetti A, Fraggetta F, Terzi A, Iannucci A, Cetto GL. Immunocytochemical assessment of bone marrow aspirates for monitoring response to chemotherapy in small-cell lung cancer patients. Br J Cancer 1999;81(7):1213-1221. https://doi.org/10.1038/sj.bjc.6690831
- Myklebust AT, Pharo A, Fodstad O: Effective removal of SCLC cells from human bone marrow. Use of four monoclonal antibodies and immunomagnetic beads. Br J Cancer 1993;67(6):1331-1336. https://doi.org/10.1038/bjc.1993.246
- Chambers AF, Naumov GN, Vantyghem SA, Tuck AB: Molecular biology of breast cancer metastasis. Clinical implications of experimental studies on metastatic inefficiency. Breast Cancer Res 2000;2(6):400-407. https://doi.org/10.1186/bcr86
- Guba M, Cernaianu G, Koehl G, Geissler EK, Jauch KW, Anthuber M, Falk W, Steinbauer M: A primary tumor promotes dormancy of solitary tumor cells before inhibiting angiogenesis. Cancer Res 2001;61(14):5575-5579. PMID: 11454710.
- Naumov GN, MacDonald IC, Chambers AF, Groom AC: Solitary cancer cells as a possible source of tumour dormancy? Semin Cancer Biol 2001;11(4):271-276. https://doi.org/10.1006/scbi.2001.0382
- Aguirre-Ghiso JA: Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 2007;7(11):834-846. https://doi.org/10.1038/nrc2256
- Sosa MS, Bragado P, Aguirre-Ghiso JA: Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 2014;14(9):611-622. https://doi.org/10.1038/nrc3793
- Zhang J, Zhang J, Han L, Wu S, Li J, Eaton EN, Yuan B, Reinhardt F, Li H, Strasser PC, Das S, Liu Donaher J, Khalil MI, Jiang H, Deuschel A, Lin D, Sebastiany C, Maranga M, Shubitidze S, Liu X, Lambert AW, Zhang Y, Liu Y, Sui L, Elmiligy S, Pozza U, Günsay R, Mishra R, Velarde J, Iyer S, Henry WS, Weiskopf K, Feng G, Oni TE, Watnick RS, Li X, Weinberg RA: Inflammation awakens dormant cancer cells by modulating the epithelial-mesenchymal phenotypic state. Proc Natl Acad Sci USA 2025;122(36):e2515009122. https://doi.org/10.1073/pnas.2515009122
- Folkman J: Tumor angiogenesis. Adv Cancer Res 1985;43:175-203. https://doi.org/10.1016/s0065-230x(08)60946-x
- Burnet FM: The concept of immunological surveillance. Prog Exp Tumor Res 1970;13:1-27. https://doi.org/10.1159/000386035
- Eccles SA, Alexander P: Immunologically-mediated restraint of latent tumour metastases. Nature 1975;257(5521):52-53. https://doi.org/10.1038/257052a0
- Weinhold KJ, Goldstein LT, Wheelock EF: Tumour-dormant states established with L5178Y lymphoma cells in immunised syngeneic murine hosts. Nature 1977;270(5632):59-61. https://doi.org/10.1038/270059a0
- Weinhold KJ, Goldstein LT, Wheelock EF: The tumor dormant state. Quantitation of L5178Y cells and host immune responses during the establishment and course of dormancy in syngeneic DBA/2 mice. J Exp Med 1979;149(3):732-744. https://doi.org/10.1084/jem.149.3.732
- Holmgren L, O'Reilly MS, Folkman J: Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med 1995;1(2):149-153. https://doi.org/10.1038/nm0295-149
- Yu W, Kim J, Ossowski L: Reduction in surface urokinase receptor forces malignant cells into a protracted state of dormancy. J Cell Biol 1997;137(3):767-777. https://doi.org/10.1083/jcb.137.3.767
- Mazzoni E, Adam A, Bal de Kier Joffe E, Aguirre-Ghiso JA: Immortalized mammary epithelial cells overexpressing protein kinase C gamma acquire a malignant phenotype and become tumorigenic in vivo. Mol Cancer Res 2003;1(10):776-87. PMID: 12939403.
- Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L: ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res 2003;63(7):1684-95. PMID: 12670923.
- Kummer JL, Rao PK, Heidenreich KA: Apoptosis induced by withdrawal of trophic factors is mediated by p38 mitogen-activated protein kinase. J Biol Chem 1997;272(33):20490-4. https://doi.org/10.1074/jbc.272.33.20490
- Bulavin DV, Fornace AJ Jr: p38 MAP kinase's emerging role as a tumor suppressor. Adv Cancer Res 2004;92:95-118. https://doi.org/10.1016/S0065-230X(04)92005-2
- Bradham C, McClay DR. p38 MAPK in development and cancer. Cell Cycle 2006;5(8):824-828. https://doi.org/10.4161/cc.5.8.2685
- Ranganathan AC, Adam AP, Zhang L, Aguirre-Ghiso JA: Tumor cell dormancy induced by p38SAPK and ER-stress signaling: an adaptive advantage for metastatic cells? Cancer Biol Ther 2006;5(7):729-735. https://doi.org/10.4161/cbt.5.7.2968
- Cameron MD, Schmidt EE, Kerkvliet N, Nadkarni KV, Morris VL, Groom AC, Chambers AF, MacDonald IC: Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res 2000;60(9):2541-2546. PMID: 10811137.
- Naumov GN, MacDonald IC, Weinmeister PM, Kerkvliet N, Nadkarni KV, Wilson SM, Morris VL, Groom AC, Chambers AF: Persistence of solitary mammary carcinoma cells in a secondary site: a possible contributor to dormancy. Cancer Res 2002;62(7):2162-2168. PMID: 11929839.
- Naumov GN, Townson JL, MacDonald IC, Wilson SM, Bramwell VH, Groom AC, Chambers AF: Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Res Treat 2003;82(3):199-206. https://doi.org/10.1023/B:BREA.0000004377.12288.3c
- Dong Z, Kumar R, Yang X, Fidler IJ: Macrophage-derived metalloelastase is responsible for the generation of angiostatin in Lewis lung carcinoma. Cell 1997;88(6):801-810. https://doi.org/10.1016/s0092-8674(00)81926-1
- Gimbrone MA Jr, Leapman SB, Cotran RS, Folkman J: Tumor dormancy in vivo by prevention of neovascularization. J Exp Med 1972;136(2):261-276. https://doi.org/10.1084/jem.136.2.261
- Jamaluddin MS: Aspirin upregulates expression of urokinase type plasminogen activator receptor (uPAR) gene in human colon cancer cells through AP1. Biochem Biophys Res Commun 2006;348(2):618-627. https://doi.org/10.1016/j.bbrc.2006.07.098
- Aguirre Ghiso JA, Kovalski K, Ossowski L: Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol 1999;147(1):89-104. https://doi.org/10.1083/jcb.147.1.89
- Gao H, Chakraborty G, Lee-Lim AP, Mavrakis KJ, Wendel HG, Giancotti FG: Forward genetic screens in mice uncover mediators and suppressors of metastatic reactivation. Proc Natl Acad Sci USA 2014;111(46):16532-15537. https://doi.org/10.1073/pnas.1403234111
- Song F, Wei M, Wang J, Liu Y, Guo M, Li X, Luo J, Zhou J, Wang M, Guo D, Chen L, Sun G: Hepatitis B virus-regulated growth of liver cancer cells occurs through the microRNA-340-5p-activating transcription factor 7-heat shock protein A member 1B axis. Cancer Sci 2019;110(5):1633-1643. https://doi.org/10.1111/cas.14004
- Almog N, Ma L, Schwager C, Brinkmann BG, Beheshti A, Vajkoczy P, Folkman J, Hlatky L, Abdollahi A: Consensus micro RNAs governing the switch of dormant tumors to the fast-growing angiogenic phenotype. PLoS One 2012;7(8):e44001. https://doi.org/10.1371/journal.pone.0044001
- Naumov GN, Bender E, Zurakowski D, Kang SY, Sampson D, Flynn E, Watnick RS, Straume O, Akslen LA, Folkman J, Almog N: A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J Natl Cancer Inst 2006;98(5):316-325. https://doi.org/10.1093/jnci/djj068
- Barkan D, Kleinman H, Simmons JL, Asmussen H, Kamaraju AK, Hoenorhoff MJ, Liu ZY, Costes SV, Cho EH, Lockett S, Khanna C, Chambers AF, Green JE: Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res 2008;68(15):6241-6250. https://doi.org/10.1158/0008-5472.CAN-07-6849
- Kmieciak M, Payne KK, Wang XY, Manjili MH: IFN-γ Rα is a key determinant of CD8+ T cell-mediated tumor elimination or tumor escape and relapse in FVB mouse. PLoS One 2013;8(12):e82544. https://doi.org/10.1371/journal.pone.0082544
- Nicolas E, Kosmider B, Cukierman E, Borghaei H, Golemis EA, Borriello L: Cancer treatments as paradoxical catalysts of tumor awakening in the lung. Cancer Metastasis Rev 2024;43(4):1165-1183. https://doi.org/10.1007/s10555-024-10196-5
- He D, Wu Q, Tian P, Liu Y, Jia Z, Li Z, Wang Y, Jin Y, Luo W, Li L, Zhang P, Jin Q, Zhao W, Hu W, Liang Y, Zhou B, Yang Q, Jiang YZ, Shao ZM, Hu G: Chemotherapy awakens dormant cancer cells in lung by inducing neutrophil extracellular traps. Cancer Cell 2025;43(9):1622-1636.e7. https://doi.org/10.1016/j.ccell.2025.06.007
- Zhang J, Zhang J, Han L, Wu S, Li J, Eaton EN, Yuan B, Reinhardt F, Li H, Strasser PC, Das S, Liu Donaher J, Khalil MI, Jiang H, Deuschel A, Lin D, Sebastiany C, Maranga M, Shubitidze S, Liu X, Lambert AW, Zhang Y, Liu Y, Sui L, Elmiligy S, Pozza U, Günsay R, Mishra R, Velarde J, Iyer S, Henry WS, Weiskopf K, Feng G, Oni TE, Watnick RS, Li X, Weinberg RA: Inflammation awakens dormant cancer cells by modulating the epithelial-mesenchymal phenotypic state. Proc Natl Acad Sci USA 2025;122(36):e2515009122. https://doi.org/10.1073/pnas.2515009122
- Chia SB, Johnson BJ, Hu J, Valença-Pereira F, Chadeau-Hyam M, Guntoro F, Montgomery H, Boorgula MP, Sreekanth V, Goodspeed A, Davenport B, De Dominici M, Zaberezhnyy V, Schleicher WE, Gao D, Cadar AN, Petriz-Otaño L, Papanicolaou M, Beheshti A, Baylin SB, Guarnieri JW, Wallace DC, Costello JC, Bartley JM, Morrison TE, Vermeulen R, Aguirre-Ghiso JA, Rincon M, DeGregori J: Respiratory viral infections awaken metastatic breast cancer cells in lungs. Nature 2025;645(8080):496-506. https://doi.org/10.1038/s41586-025-09332-0
- Perego M, Tyurin VA, Tyurina YY, Yellets J, Nacarelli T, Lin C, Nefedova Y, Kossenkov A, Liu Q, Sreedhar S, Pass H, Roth J, Vogl T, Feldser D, Zhang R, Kagan VE, Gabrilovich DI: Reactivation of dormant tumor cells by modified lipids derived from stress-activated neutrophils. Sci Transl Med 2020;12(572):eabb5817. https://doi.org/10.1126/scitranslmed.abb5817
- Cho J, Lee HJ, Hwang SJ, Min HY, Kang HN, Park AY, Hyun SY, Sim JY, Lee HJ, Jang HJ, Suh YA, Hong S, Shin YK, Kim HR, Lee HY: The Interplay between Slow-Cycling, Chemoresistant Cancer Cells and Fibroblasts Creates a Proinflammatory Niche for Tumor Progression. Cancer Res 2020;80(11):2257-2272. https://doi.org/10.1158/0008-5472.CAN-19-0631
- Jo H, Jia Y, Subramanian KK, Hattori H, Luo HR: Cancer cell-derived clusterin modulates the phosphatidylinositol 3'-kinase-Akt pathway through attenuation of insulin-like growth factor 1 during serum deprivation. Mol Cell Biol 2008;28(13):4285-4299. https://doi.org/10.1128/MCB.01240-07
- Schewe DM, Aguirre-Ghiso JA: ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci USA 2008;105(30):10519-10524. https://doi.org/10.1073/pnas.0800939105
- Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S, Pai SK, Liu W, Fukuda K, Chambers C, Wilber A, Watabe K: Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med 2011;208(13):2641-2655. https://doi.org/10.1084/jem.20110840
- Zhang WL, Fan HY, Chen BJ, Wang HF, Pang X, Li M, Liang XH, Tang YL: Cancer-associated fibroblasts-derived CXCL1 activates DEC2-mediated dormancy in oral squamous cell carcinoma. Heliyon 2024;10(20):e39133. https://doi.org/10.1016/j.heliyon.2024.e39133
- Liang Y, Chen WM, Zhang Y, Li L: Remodeling the tumor dormancy ecosystem to prevent recurrence and metastasis. Signal Transduct Target Ther 2026;11(1):1. https://doi.org/10.1038/s41392-025-02328-2
- Pereira P, Panier J, Nater M, Herbst M, Calvanese AL, Köksal H, Castañón H, Cecconi V, Tallón de Lara P, Pascolo S, van den Broek M: Inflammatory cytokines mediate the induction of and awakening from metastatic dormancy. Cell Rep 2025;44(3):115388. https://doi.org/10.1016/j.celrep.2025.115388
- Yilmaz A, Haerri L, Granda ME, Coquoz O, Lan Q, Rüegg C: The CXCL10/CXCR3 axis is essential for sustaining immunological dormancy in triple-negative breast cancer. NPJ Breast Cancer 2026;12(1):36. https://doi.org/10.1038/s41523-026-00903-6
- Nobre AR, Risson E, Singh DK, Di Martino JS, Cheung JF, Wang J, Johnson J, Russnes HG, Bravo-Cordero JJ, Birbrair A, Naume B, Azhar M, Frenette PS, Aguirre-Ghiso JA: Bone marrow NG2+/Nestin+ mesenchymal stem cells drive DTC dormancy via TGFβ2. Nat Cancer 2021;2(3):327-339. https://doi.org/10.1038/s43018-021-00179-8
- Singh DK, Carcamo S, Farias EF, Hasson D, Zheng W, Sun D, Huang X, Cheung J, Nobre AR, Kale N, Sosa MS, Bernstein E, Aguirre-Ghiso JA: 5-Azacytidine- and retinoic-acid-induced reprogramming of DCCs into dormancy suppresses metastasis via restored TGF-β-SMAD4 signaling. Cell Rep 2023;42(6):112560. https://doi.org/10.1016/j.celrep.2023.112560
- Xiao ZQ, Majumdar AP: Induction of transcriptional activity of AP-1 and NF-kappaB in the gastric mucosa during aging. Am J Physiol Gastrointest Liver Physiol 2000;278(6):G855-65. https://doi.org/10.1152/ajpgi.2000.278.6.G855
- Fujioka S, Niu J, Schmidt C, Sclabas GM, Peng B, Uwagawa T, Li Z, Evans DB, Abbruzzese JL, Chiao PJ: NF-kappaB and AP-1 connection: mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol Cell Biol 2004;24(17):7806-7819. https://doi.org/10.1128/MCB.24.17.7806-7819.2004
- Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, Upadhyay P, Uyeminami DL, Pommier A, Küttner V, Bružas E, Maiorino L, Bautista C, Carmona EM, Gimotty PA, Fearon DT, Chang K, Lyons SK, Pinkerton KE, Trotman LC, Goldberg MS, Yeh JT, Egeblad M: Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018;361(6409):eaao4227. https://doi.org/10.1126/science.aao4227
- Hoppe-Seyler K, Bossler F, Lohrey C, Bulkescher J, Rösl F, Jansen L, Mayer A, Vaupel P, Dürst M, Hoppe-Seyler F: Induction of dormancy in hypoxic human papillomavirus-positive cancer cells. Proc Natl Acad Sci USA 2017;114(6):E990-E998. https://doi.org/10.1073/pnas.1615758114
- Liu Y, Ao X, Ding W, Ponnusamy M, Wu W, Hao X, Yu W, Wang Y, Li P, Wang J: Critical role of FOXO3a in carcinogenesis. Mol Cancer 2018;17(1):104. https://doi.org/10.1186/s12943-018-0856-3
- Saatcioglu HD, Cuevas I, Castrillon DH: Control of Oocyte Reawakening by Kit. PLoS Genet 2016;12(8):e1006215. https://doi.org/10.1371/journal.pgen.1006215
- Vo KCT, Kawamura K: Ovarian Fragmentation and AKT Stimulation for Expansion of Fertile Lifespan. Front Reprod Health 2021;3:636771. https://doi.org/10.3389/frph.2021.636771
- Chen CC, Jeon SM, Bhaskar PT, Nogueira V, Sundararajan D, Tonic I, Park Y, Hay N: FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Dev Cell 2010;18(4):592-604. https://doi.org/10.1016/j.devcel.2010.03.008
- Liu Y, Azizian NG, Sullivan DK, Li Y: mTOR inhibition attenuates chemosensitivity through the induction of chemotherapy resistant persisters. Nat Commun 2022;13(1):7047. https://doi.org/10.1038/s41467-022-34890-6
- Aleksandrova KV, Vorobev ML, Suvorova II: mTOR pathway occupies a central role in the emergence of latent cancer cells. Cell Death Dis 2024;15(2):176. https://doi.org/10.1038/s41419-024-06547-3
- Fan C, Zhao C, Zhang F, Kesarwani M, Tu Z, Cai X, Davis AK, Xu L, Hochstetler CL, Chen X, Guo F, Huang G, Azam M, Tian W, Lu QR, Zheng Y: Adaptive responses to mTOR gene targeting in hematopoietic stem cells reveal a proliferative mechanism evasive to mTOR inhibition. Proc Natl Acad Sci USA 2021;118(1):e2020102118. https://doi.org/10.1073/pnas.2020102118
- Qiu H, Orr FW, Jensen D, Wang HH, McIntosh AR, Hasinoff BB, Nance DM, Pylypas S, Qi K, Song C, Muschel RJ, Al-Mehdi AB: Arrest of B16 melanoma cells in the mouse pulmonary microcirculation induces endothelial nitric oxide synthase-dependent nitric oxide release that is cytotoxic to the tumor cells. Am J Pathol 2003;162(2):403-412.: https://doi.org/10.1016/S0002-9440(10)63835-7
- Fukumura D, Kashiwagi S, Jain RK: The role of nitric oxide in tumour progression. Nat Rev Cancer 2006;6(7):521-534. https://doi.org/10.1038/nrc1910
- Tsai NM, Chen BM, Wei SL, Wu CW, Roffler SR: Anti-tumor immunoglobulin M increases lung metastasis in an experimental model of malignant melanoma. Clin Exp Metastasis 2003;20(2):103-109. https://doi.org/10.1023/a:1022616223359
- Nagashima R, Maeda K, Imai Y, Takahashi T: Lamina propria macrophages in the human gastrointestinal mucosa: their distribution, immunohistological phenotype, and function. J Histochem Cytochem 1996;44(7):721-731. https://doi.org/10.1177/44.7.8675993
- Garrido F, Ruiz-Cabello F, Cabrera T, Pérez-Villar JJ, López-Botet M, Duggan-Keen M, Stern PL: Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol Today 1997;18(2):89-95. https://doi.org/10.1016/s0167-5699(96)10075-x
- Ikeda H, Lethé B, Lehmann F, van Baren N, Baurain JF, de Smet C, Chambost H, Vitale M, Moretta A, Boon T, Coulie PG: Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity 1997;6(2):199-208. https://doi.org/10.1016/s1074-7613(00)80426-4
- Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, Marshall JF, Chin-Aleong J, Chelala C, Gribben JG, Ramsay AG, Kocher HM: Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013;145(5):1121-1132. https://doi.org/10.1053/j.gastro.2013.07.025
- Karlhofer FM, Ribaudo RK, Yokoyama WM: MHC class I alloantigen specificity of Ly-49+ IL-2-activated natural killer cells. Nature 1992;358(6381):66-70. https://doi.org/10.1038/358066a0
- Juste-Lanas Y, Díaz-Valdivia N, Llorente A, Ikemori R, Bernardo A, Arshakyan M, Borau C, Ramírez J, Ruffinelli JC, Nadal E, Reguart N, García-Aznar JM, Alcaraz J: 3D collagen migration patterns reveal a SMAD3-dependent and TGF-β1-independent mechanism of recruitment for tumour-associated fibroblasts in lung adenocarcinoma, Br J Cancer 2023;128(6):967-981. https://doi.org/10.1038/s41416-022-02093-x
- Fullár A, Dudás J, Oláh L, Hollósi P, Papp Z, Sobel G, Karászi K, Paku S, Baghy K, Kovalszky I: Remodeling of extracellular matrix by normal and tumor-associated fibroblasts promotes cervical cancer progression. BMC Cancer 2015;15:256. https://doi.org/10.1186/s12885-015-1272-3
- Kinoshita T, Ishii G, Hiraoka N, Hirayama S, Yamauchi C, Aokage K, Hishida T, Yoshida J, Nagai K, Ochiai A: Forkhead box P3 regulatory T cells coexisting with cancer associated fibroblasts are correlated with a poor outcome in lung adenocarcinoma. Cancer Sci 2013;104(4):409-415. https://doi.org/10.1111/cas.12099
- Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, Odze R, Glickman JN, Garrett WS: CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep 2015;12(2):244-257. https://doi.org/10.1016/j.celrep.2015.06.024
- Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G: Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med 2014;20(6):607-615. https://doi.org/10.1038/nm.3541
- Györy I, Boller S, Nechanitzky R, Mandel E, Pott S, Liu E, Grosschedl R: Transcription factor Ebf1 regulates differentiation stage-specific signaling, proliferation, and survival of B cells. Genes Dev 2012;26(7):668-682. https://doi.org/10.1101/gad.187328.112
- Li JP, Liu YJ, Wang SS, Lu ZH, Ye QW, Zhou JY, Zou X, Chen YG: EBF1-COX4I2 signaling axis promotes a myofibroblast-like phenotype in cancer-associated fibroblasts (CAFs) and is associated with an immunosuppressive microenvironment. Int Immunopharmacol 2024;139:112666. https://doi.org/10.1016/j.intimp.2024.112666
- Lagergren A, Månsson R, Zetterblad J, Smith E, Basta B, Bryder D, Akerblad P, Sigvardsson M: The Cxcl12, periostin, and Ccl9 genes are direct targets for early B-cell factor in OP-9 stroma cells. J Biol Chem. 2007 May 11;282(19):14454-14462. https://doi.org/10.1074/jbc.M610263200
- Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK: Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity 2002;16(2):219-230. https://doi.org/10.1016/s1074-7613(02)00274-1
- Pillai S, Mattoo H, Cariappa A: B cells and autoimmunity. Curr Opin Immunol 2011;23(6):721-731. doi: https://doi.org/10.1016/j.coi.2011.10.007
- Inoue S, Leitner WW, Golding B, Scott D: Inhibitory effects of B cells on antitumor immunity. Cancer Res 2006;66(15):7741-7747. https://doi.org/10.1158/0008-5472.CAN-05-3766
- Shimabukuro-Vornhagen A, Schlößer HA, Gryschok L, Malcher J, Wennhold K, Garcia-Marquez M, Herbold T, Neuhaus LS, Becker HJ, Fiedler A, Scherwitz P, Koslowsky T, Hake R, Stippel DL, Hölscher AH, Eidt S, Hallek M, Theurich S, von Bergwelt-Baildon MS: Characterization of tumor-associated B-cell subsets in patients with colorectal cancer. Oncotarget 2014;5(13):4651-4664. https://doi.org/10.18632/oncotarget.1701
- Li W, Song D, Li H, Liang L, Zhao N, Liu T: Reduction in Peripheral CD19+CD24hCD27+ B Cell Frequency Predicts Favourable Clinical Course in XELOX-Treated Patients with Advanced Gastric Cancer. Cell Physiol Biochem. 2017;41(5):2045-2052. https://doi.org/10.1159/000475435
- Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA: Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007;449(7162):557-563. 10.1038/nature06188
- Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, Luu T, Li AX, Wu X, Ye W, Chen S, Zhou W, Yu Y, Wang YZ, Ren X, Li H, Scherle P, Kuroki Y, Wang SE: CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res 2012;72(11):2768-2779. https://doi.org/10.1158/0008-5472.CAN-11-3567
- Cho H, Seo Y, Loke KM, Kim SW, Oh SM, Kim JH, Soh J, Kim HS, Lee H, Kim J, Min JJ, Jung DW, Williams DR: Cancer-Stimulated CAFs Enhance Monocyte Differentiation and Protumoral TAM Activation via IL6 and GM-CSF Secretion. Clin Cancer Res 2018;24(21):5407-5421. https://doi.org/10.1158/1078-0432.CCR-18-0125
- Fang WB, Jokar I, Zou A, Lambert D, Dendukuri P, Cheng N: CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms. J Biol Chem 2012;287(43):36593-36608. https://doi.org/10.1074/jbc.M112.365999
- Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, Odze R, Glickman JN, Garrett WS: CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep 2015;12(2):244-257. https://doi.org/10.1016/j.celrep.2015.06.024
- Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, Zhang B, Meng Q, Yu X, Shi S: Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer 2021;20(1):131. https://doi.org/10.1186/s12943-021-01428-1
- Baldominos P, Barbera-Mourelle A, Barreiro O, Huang Y, Wight A, Cho JW, Zhao X, Estivill G, Adam I, Sanchez X, McCarthy S, Schaller J, Khan Z, Ruzo A, Pastorello R, Richardson ET, Dillon D, Montero-Llopis P, Barroso-Sousa R, Forman J, Shukla SA, Tolaney SM, Mittendorf EA, von Andrian UH, Wucherpfennig KW, Hemberg M, Agudo J: Quiescent cancer cells resist T cell attack by forming an immunosuppressive niche. Cell 2022;185(10):1694-1708.e19. https://doi.org/10.1016/j.cell.2022.03.033
- Garcia-Recio S, Hinoue T, Wheeler GL, Kelly BJ, Garrido-Castro AC, Pascual T, De Cubas AA, Xia Y, Felsheim BM, McClure MB, Rajkovic A, Karaesmen E, Smith MA, Fan C, Ericsson PIG, Sanders ME, Creighton CJ, Bowen J, Leraas K, Burns RT, Coppens S, Wheless A, Rezk S, Garrett AL, Parker JS, Foy KK, Shen H, Park BH, Krop I, Anders C, Gastier-Foster J, Rimawi MF, Nanda R, Lin NU, Isaacs C, Marcom PK, Storniolo AM, Couch FJ, Chandran U, Davis M, Silverstein J, Ropelewski A, Liu MC, Hilsenbeck SG, Norton L, Richardson AL, Symmans WF, Wolff AC, Davidson NE, Carey LA, Lee AV, Balko JM, Hoadley KA, Laird PW, Mardis ER, King TA; AURORA US Network; Perou CM: Multiomics in primary and metastatic breast tumors from the AURORA US network finds microenvironment and epigenetic drivers of metastasis, Nat Cancer 2022 Dec 30. https://doi.org/10.1038/s43018-022-00491-x
- Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, Ohtani H: CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res 1998;58(16):3491-3494 PMID: 9721846.
- Cho Y, Miyamoto M, Kato K, Fukunaga A, Shichinohe T, Kawarada Y, Hida Y, Oshikiri T, Kurokawa T, Suzuoki M, Nakakubo Y, Hiraoka K, Murakami S, Shinohara T, Itoh T, Okushiba S, Kondo S, Katoh H: CD4+ and CD8+ T Cells Cooperate to Improve Prognosis of Patients with Esophageal Squamous Cell Carcinoma. Cancer Res 2003;63(7):1555-1559 PMID: 12670904.
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM: Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 2008;322(5904):1097-2100. https://doi.org/10.1126/science.1164206
- Yin HL, Stossel TP: Control of cytoplasmic actin gel-sol transformation by gelsolin, a calcium-dependent regulatory protein. Nature 1979;281(5732):583-6. https://doi.org/10.1038/281583a0
- Yin HL, Stossel TP: Purification and structural properties of gelsolin, a Ca2+-activated regulatory protein of macrophages. J Biol Chem 1980;255(19):9490-3 PMID: 6251090.
- Yin HL, Zaner KS, Stossel TP: Ca2+ control of actin gelation. Interaction of gelsolin with actin filaments and regulation of actin gelation. J Biol Chem 1980;255(19):9494-500. PMID: 6251091.
- Ohtsu M, Sakai N, Fujita H, Kashiwagi M, Gasa S, Shimizu S, Eguchi Y, Tsujimoto Y, Sakiyama Y, Kobayashi K, Kuzumaki N: Inhibition of apoptosis by the actin-regulatory protein gelsolin. EMBO J 1997;16(15):4650-6. https://doi.org/10.1093/emboj/16.15.4650
- Porter RM, Holme TC, Newman EL, Hopwood D, Wilkinson JM, Cuschieri A: Monoclonal antibodies to cytoskeletal proteins: an immunohistochemical investigation of human colon cancer. J Pathol 1993;170(4):435-40. https://doi.org/10.1002/path.1711700406
- Asch HL, Head K, Dong Y, Natoli F, Winston JS, Connolly JL, Asch BB: Widespread loss of gelsolin in breast cancers of humans, mice, and rats. Cancer Res 1996;56(21):4841-4845 PMID: 8895730.
- Mielnicki LM, Ying AM, Head KL, Asch HL, Asch BB: Epigenetic regulation of gelsolin expression in human breast cancer cells. Exp Cell Res 1999;249(1):161-76. https://doi.org/10.1006/excr.1999.4461
- Lee HK, Driscoll D, Asch H, Asch B, Zhang PJ: Downregulated gelsolin expression in hyperplastic and neoplastic lesions of the prostate. Prostate 1999;40(1):14-19. https://doi.org/10.1002/(sici)1097-0045(19990615)40:1<14::aid-pros2>3.0.co;2-6
- Wang Z, Elbanna Y, Godet I, Gan S, Peters L, Lampe G, Chen Y, Xavier J, Huse M, Massagué J: TGFbeta induces an atypical EMT to evade immune mechanosurveillance in lung adenocarcinoma dormant metastasis. Nat Cancer 2026 Jan 5. https://doi.org/10.1038/s43018-025-01094-y
- Swamydas M, Nguyen D, Allen LD, Eddy J, Dréau D: Progranulin stimulated by LPA promotes the migration of aggressive breast cancer cells. Cell Commun Adhes 2011;18(6):119-130. https://doi.org/10.3109/15419061.2011.641042
- Wang H, Sun Y, Liu S, Yu H, Li W, Zeng J, Chen C, Jia J: Upregulation of progranulin by Helicobacter pylori in human gastric epithelial cells via p38MAPK and MEK1/2 signaling pathway: role in epithelial cell proliferation and migration. FEMS Immunol Med Microbiol 2011;63(1):82-92. https://doi.org/10.1111/j.1574-695X.2011.00833.x
- Jones MB, Michener CM, Blanchette JO, Kuznetsov VA, Raffeld M, Serrero G, Emmert-Buck MR, Petricoin EF, Krizman DB, Liotta LA, Kohn EC: The granulin-epithelin precursor/PC-cell-derived growth factor is a growth factor for epithelial ovarian cancer. Clin Cancer Res 2003;9(1):44-51. PMID: 12538450.
- Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, Ireland L, Sakai T, Sakai K, Kim YS, Engle D, Campbell F, Palmer D, Ko JH, Tuveson DA, Hirsch E, Mielgo A, Schmid MC: Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol 2016;18(5):549-560. https://doi.org/10.1038/ncb3340 Corrigendum: https://doi.org/10.1038/ncb3377
- Liu L, Xiang M, Zhou J, Ren Z, Shi W, Du X, Fu X, Li P, Wang H: Progranulin inhibits autophagy to facilitate intracellular colonization of Helicobacter pylori through the PGRN/mTOR/DCN axis in gastric epithelial cells. Front Cell Infect Microbiol 2024;14:1425367. https://doi.org/10.3389/fcimb.2024.1425367
- Pan Y, Cheung ST, Tong JHM, Tin KY, Kang W, Lung RWM, Wu F, Li H, Ng SSM, Mak TWC, To KF, Chan AWH: Granulin epithelin precursor promotes colorectal carcinogenesis by activating MARK/ERK pathway. J Transl Med 2018;16(1):150. https://doi.org/10.1186/s12967-018-1530-7
- Zhao J, Li X, Liu J, Jiang W, Wen D, Xue H: Effect of Progranulin on Migration and Invasion of Human Colon Cancer Cells. J Coll Physicians Surg Pak 2018;28(8):607-611. https://doi.org/10.29271/jcpsp.2018.08.607
- Chen R, Zhang X, Li B, Tonetti MS, Yang Y, Li Y, Liu B, Qian S, Gu Y, Wang Q, Mao K, Cheng H, Lai H, Shi J: Progranulin-dependent repair function of regulatory T cells drives bone-fracture healing. J Clin Invest 2024;135(2):e180679. https://doi.org/10.1172/JCI180679
- Moresco P, Kastan JP, Yang JI, Prabakar R, Kelley ZL, Minicozzi F, Adams DW, Cifani P, Tuveson DA, Fearon DT: Signal peptide-independent secretion of keratin-19 by pancreatic cancer cells. Proc Natl Acad Sci USA 2025;122(27):e2426218122. https://doi.org/10.1073/pnas.2426218122
- Schoenfeld A, Luqmani Y, Sinnett HD, Shousha S, Coombes RC: Keratin 19 mRNA measurement to detect micrometastases in lymph nodes in breast cancer patients. Br J Cancer 1996;74(10):1639-1642. https://doi.org/10.1038/bjc.1996.601
- Fallatah A, Anastasakis DG, Manzourolajdad A, Sharma P, Wang X, Jacob A, Alsharif S, Elgerbi A, Coulombe PA, Hafner M, Chung BM: Keratin 19 binds and regulates cytoplasmic HNRNPK mRNA targets in triple-negative breast cancer. BMC Mol Cell Biol 2023;24(1):26. https://doi.org/10.1186/s12860-023-00488-z
- Shi HQ, Li X, Chen Z, Dong S, Ye C, Hou S, Fan DA, Zhang H, Zhou WC: KRT19 is regulated by miR-642a-5p and promotes pancreatic cancer progression through the Wnt/β-catenin pathway. iScience 2024;27(9):110782. https://doi.org/10.1016/j.isci.2024.110782
- Alsharif S, Sharma P, Bursch K, Milliken R, Lam V, Fallatah A, Phan T, Collins M, Dohlman P, Tiufekchiev S, Nehmetallah G, Raub CB, Chung BM: Keratin 19 maintains E-cadherin localization at the cell surface and stabilizes cell-cell adhesion of MCF7 cells. Cell Adh Migr 2021;15(1):1-17. https://doi.org/10.1080/19336918.2020.1868694
- Uenishi T, Kubo S, Yamamoto T, Shuto T, Ogawa M, Tanaka H, Tanaka S, Kaneda K, Hirohashi K: Cytokeratin 19 expression in hepatocellular carcinoma predicts early postoperative recurrence. Cancer Sci 2003;94(10):851-857. https://doi.org/10.1111/j.1349-7006.2003.tb01366.x
- Kim H, Choi GH, Na DC, Ahn EY, Kim GI, Lee JE, Cho JY, Yoo JE, Choi JS, Park YN: Human hepatocellular carcinomas with "Stemness"-related marker expression: keratin 19 expression and a poor prognosis. Hepatology 2011;54(5):1707-1717. https://doi.org/10.1002/hep.24559
- Takano M, Shimada K, Fujii T, Morita K, Takeda M, Nakajima Y, Nonomura A, Konishi N, Obayashi C: Keratin 19 as a key molecule in progression of human hepatocellular carcinomas through invasion and angiogenesis. BMC Cancer 2016;16(1):903. https://doi.org/10.1186/s12885-016-2949-y
- Takeshita S, Kikuno R, Tezuka K, Amann E: Osteoblast-specific factor 2: cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem J 1993;294 ( Pt 1)(Pt 1):271-278. https://doi.org/10.1042/bj2940271
- Norris RA, Damon B, Mironov V, Kasyanov V, Ramamurthi A, Moreno-Rodriguez R, Trusk T, Potts JD, Goodwin RL, Davis J, Hoffman S, Wen X, Sugi Y, Kern CB, Mjaatvedt CH, Turner DK, Oka T, Conway SJ, Molkentin JD, Forgacs G, Markwald RR: Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem 2007;101(3):695-711. https://doi.org/10.1002/jcb.21224
- Gadermaier E, Tesarz M, Suciu AA, Wallwitz J, Berg G, Himmler G: Characterization of a sandwich ELISA for the quantification of all human periostin isoforms. J Clin Lab Anal 2018;32(2):e22252. https://doi.org/10.1002/jcla.22252
- Gillan L, Matei D, Fishman DA, Gerbin CS, Karlan BY, Chang DD: Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res 2002;62(18):5358-64. PMID: 12235007.
- Hu Q, Tong S, Zhao X, Ding W, Gou Y, Xu K, Sun C, Xia G: Periostin Mediates TGF-beta-Induced Epithelial Mesenchymal Transition in Prostate Cancer Cells. Cell Physiol Biochem 2015;36(2):799-809. https://doi.org/10.1159/000430139
- Sasaki H, Lo KM, Chen LB, Auclair D, Nakashima Y, Moriyama S, Fukai I, Tam C, Loda M, Fujii Y: Expression of Periostin, homologous with an insect cell adhesion molecule, as a prognostic marker in non-small cell lung cancers. Jpn J Cancer Res 2001;92(8):869-73. https://doi.org/10.1111/j.1349-7006.2001.tb01174.x
- Michaylira CZ, Wong GS, Miller CG, Gutierrez CM, Nakagawa H, Hammond R, Klein-Szanto AJ, Lee JS, Kim SB, Herlyn M, Diehl JA, Gimotty P, Rustgi AK: Periostin, a cell adhesion molecule, facilitates invasion in the tumor microenvironment and annotates a novel tumor-invasive signature in esophageal cancer. Cancer Res 2010;70(13):5281-5292. https://doi.org/10.1158/0008-5472.CAN-10-0704
- Underwood TJ, Hayden AL, Derouet M, Garcia E, Noble F, White MJ, Thirdborough S, Mead A, Clemons N, Mellone M, Uzoho C, Primrose JN, Blaydes JP, Thomas GJ: Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J Pathol 2015;235(3):466-477. https://doi.org/10.1002/path.4467
- Gonzalez HE, Gujrati M, Frederick M, Henderson Y, Arumugam J, Spring PW, Mitsudo K, Kim HW, Clayman GL: Identification of 9 genes differentially expressed in head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg 2003;129(7):754-759. https://doi.org/10.1001/archotol.129.7.754
- Tai IT, Dai M, Chen LB: Periostin induction in tumor cell line explants and inhibition of in vitro cell growth by anti-periostin antibodies. Carcinogenesis 2005;26(5):908-915. https://doi.org/10.1093/carcin/bgi034
- Kudo Y, Siriwardena BS, Hatano H, Ogawa I, Takata T: Periostin: novel diagnostic and therapeutic target for cancer. Histol Histopathol 2007;22(10):1167-1174. https://doi.org/10.14670/HH-22.1167
- Kozono S, Ohuchida K, Eguchi D, Ikenaga N, Fujiwara K, Cui L, Mizumoto K, Tanaka M: Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res 2013;73(7):2345-2356. https://doi.org/10.1158/0008-5472.CAN-12-3180
- Maruhashi T, Kii I, Saito M, Kudo A: Interaction between periostin and BMP-1 promotes proteolytic activation of lysyl oxidase. J Biol Chem 2010;285(17):13294-13303. https://doi.org/10.1074/jbc.M109.088864
- Sun CY, Mi YY, Ge SY, Hu QF, Xu K, Guo YJ, Tan YF, Zhang Y, Zhong F, Xia GW: Tumor- and Osteoblast-Derived Periostin in Prostate Cancer bone Metastases. Front Oncol 2022;11:795712. https://doi.org/10.3389/fonc.2021.795712
- Massy E, Rousseau JC, Gueye M, Bonnelye E, Brevet M, Chambard L, Duruisseaux M, Borel O, Roger C, Guelminger R, Pialat JB, Gineyts E, Bouazza L, Millet M, Maury JM, Clézardin P, Girard N, Confavreux CB: Serum total periostin is an independent marker of overall survival in bone metastases of lung adenocarcinoma. J Bone Oncol 2021;29:100364. https://doi.org/10.1016/j.jbo.2021.100364
- Sasaki H, Yu CY, Dai M, Tam C, Loda M, Auclair D, Chen LB, Elias A: Elevated serum periostin levels in patients with bone metastases from breast but not lung cancer. Breast Cancer Res Treat 2003;77(3):245-252. https://doi.org/10.1023/a:1021899904332
- Contié S, Voorzanger-Rousselot N, Litvin J, Clézardin P, Garnero P: Increased expression and serum levels of the stromal cell-secreted protein periostin in breast cancer bone metastases. Int J Cancer 2011;128(2):352-360. https://doi.org/10.1002/ijc.25591
- Kharaishvili G, Cizkova M, Bouchalova K, Mgebrishvili G, Kolar Z, Bouchal J: Collagen triple helix repeat containing 1 protein, periostin and versican in primary and metastatic breast cancer: an immunohistochemical study. J Clin Pathol 2011;64(11):977-982. https://doi.org/10.1136/jclinpath-2011-200106
- Tian B, Zhang Y, Li N, Liu X, Dong J: CD74: a potential novel target for triple-negative breast cancer. Tumour Biol 2012 Dec;33(6):2273-2277. https://doi.org/10.1007/s13277-012-0489-x
- Gineyts E, Bonnet N, Bertholon C, Millet M, Pagnon-Minot A, Borel O, Geraci S, Bonnelye E, Croset M, Suhail A, Truica C, Lamparella N, Leitzel K, Hartmann D, Chapurlat R, Lipton A, Garnero P, Ferrari S, Clézardin P, Rousseau JC: The C-Terminal Intact Forms of Periostin (iPTN) Are Surrogate Markers for Osteolytic Lesions in Experimental Breast Cancer Bone Metastasis. Calcif Tissue Int 2018;103(5):567-580. https://doi.org/10.1007/s00223-018-0444-y
- Kumar P, Smith T, Raeman R, Chopyk DM, Brink H, Liu Y, Sulchek T, Anania FA: Periostin promotes liver fibrogenesis by activating lysyl oxidase in hepatic stellate cells. J Biol Chem 2018;293(33):12781-12792. https://doi.org/10.1074/jbc.RA117.001601
- Liu B, Wu T, Lin B, Liu X, Liu Y, Song G, Fan C, Ouyang G: Periostin-TGF-β feedforward loop contributes to tumour-stroma crosstalk in liver metastatic outgrowth of colorectal cancer. Br J Cancer 2024;130(3):358-368. https://doi.org/10.1038/s41416-023-02516-3
- Lou L, Peng K, Ouyang S, Ding W, Mo J, Yan J, Gong X, Liu G, Lu J, Yue P, Zhang K, Zhang J, Wang YD, Zhang XL: Periostin-mediated NOTCH1 activation between tumor cells and HSCs crosstalk promotes liver metastasis of small cell lung cancer. J Exp Clin Cancer Res 2025;44(1):6. https://doi.org/10.1186/s13046-024-03266-7
- Liu X, Li J, Yang X, Li X, Kong J, Qi D, Zhang F, Sun B, Liu Y, Liu T: Carcinoma-associated fibroblast-derived lysyl oxidase-rich extracellular vesicles mediate collagen crosslinking and promote epithelial-mesenchymal transition via p-FAK/p-paxillin/YAP signaling. Int J Oral Sci 2023;15(1):32. https://doi.org/10.1038/s41368-023-00236-1
- Kong J, Tian H, Zhang F, Zhang Z, Li J, Liu X, Li X, Liu J, Li X, Jin D, Yang X, Sun B, Guo T, Luo Y, Lu Y, Lin B, Liu T: Extracellular vesicles of carcinoma-associated fibroblasts creates a pre-metastatic niche in the lung through activating fibroblasts. Mol Cancer 2019;18(1):175. https://doi.org/10.1186/s12943-019-1101-4
- Kii I, Nishiyama T, Kudo A: Periostin promotes secretion of fibronectin from the endoplasmic reticulum. Biochem Biophys Res Commun. 2016;470(4):888-893. https://doi.org/10.1016/j.bbrc.2016.01.139
- Nikoloudaki G, Snider P, Simmons O, Conway SJ, Hamilton DW: Periostin and matrix stiffness combine to regulate myofibroblast differentiation and fibronectin synthesis during palatal healing. Matrix Biol 2020 Dec;94:31-56. https://doi.org/10.1016/j.matbio.2020.07.002
- Wei WF, Chen XJ, Liang LJ, Yu L, Wu XG, Zhou CF, Wang ZC, Fan LS, Hu Z, Liang L, Wang W: Periostin+ cancer-associated fibroblasts promote lymph node metastasis by impairing the lymphatic endothelial barriers in cervical squamous cell carcinoma. Mol Oncol 2021;15(1):210-227. https://doi.org/10.1002/1878-0261.12837
- Kim SS, Nikoloudaki GE, Michelsons S, Creber K, Hamilton DW: Fibronectin synthesis, but not α-smooth muscle expression, is regulated by periostin in gingival healing through FAK/JNK signaling. Sci Rep 2019;9(1):2708. https://doi.org/10.1038/s41598-018-35805-6
- Ward KA, Li W-I, Zimmer S, Davis T: Viscoelastic properties of transformed cells: role of tumor cell progression and metastasis formation. Biorheology 1991;28:301–313. https://doi.org/10.3233/bir-1991-283-419
- Waddington CH: The Strategy of Genes. A discussion of some aspects of theoretical biology. Ruskin House George Allen & Unwin Ltd., Museum Street, London. 1957.
- Gurdon JB: The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J Embryol Exp Morphol. 1962 Dec;10:622-640. https://doi.org/10.1242/dev.10.4.622
- Takahashi K, Yamanaka S: Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126(4):663-676. https://doi.org/10.1016/j.cell.2006.07.024
- Hay ED: An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 1995;154(1):8-20 https://doi.org/10.1159/000147748
- Hall PA, Lemoine NR: Rapid acinar to ductal transdifferentiation in cultured human exocrine pancreas. J Pathol 1992;166(2):97-103. https://doi.org/10.1002/path.1711660203
- Cinti S: Adipocyte differentiation and transdifferentiation: plasticity of the adipose organ. J Endocrinol Invest 2002 Nov;25(10):823-835. https://doi.org/10.1007/BF03344046
- Frid MG, Kale VA, Stenmark KR: Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ Res 2002;90(11):1189-1196. https://doi.org/10.1161/01.res.0000021432.70309.28
- Markwald RR, Fitzharris TP, Manasek FJ: Structural development of endocardial cushions. Am J Anat 1977;148(1):85-119. https://doi.org/10.1002/aja.1001480108
- Lipton BH, Bensch KG, Karasek MA: Microvessel endothelial cell transdifferentiation: phenotypic characterization. Differentiation 1991;46(2):117-133. https://doi.org/10.1111/j.1432-0436.1991.tb00872.x
- DeRuiter MC, Poelmann RE, VanMunsteren JC, Mironov V, Markwald RR, Gittenberger-de Groot AC: Embryonic endothelial cells transdifferentiate into mesenchymal cells expressing smooth muscle actins in vivo and in vitro. Circ Res 1997;80(4):444-451. https://doi.org/10.1161/01.res.80.4.444
- Condorelli G, Borello U, De Angelis L, Latronico M, Sirabella D, Coletta M, Galli R, Balconi G, Follenzi A, Frati G, Cusella De Angelis MG, Gioglio L, Amuchastegui S, Adorini L, Naldini L, Vescovi A, Dejana E, Cossu G: Cardiomyocytes induce endothelial cells to trans-differentiate into cardiac muscle: implications for myocardium regeneration. Proc Natl Acad Sci USA 2001;98(19):10733-10738. https://doi.org/10.1073/pnas.191217898
- Arciniegas E, Parada D, Graterol A: Mechanically altered embryonic chicken endothelial cells change their phenotype to an epithelioid phenotype. Anat Rec A Discov Mol Cell Evol Biol 2003;270(1):67-81 https://doi.org/10.1002/ar.a.10177
- Fujiyama S, Amano K, Uehira K, Yoshida M, Nishiwaki Y, Nozawa Y, Jin D, Takai S, Miyazaki M, Egashira K, Imada T, Iwasaka T, Matsubara H: Bone marrow monocyte lineage cells adhere on injured endothelium in a monocyte chemoattractant protein-1-dependent manner and accelerate reendothelialization as endothelial progenitor cells. Circ Res 2003;93(10):980-899. https://doi.org/10.1161/01.RES.0000099245.08637.CE
- Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R: Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 2007;67(21):10123-10128. https://doi.org/10.1158/0008-5472.CAN-07-3127
- Willis BC, duBois RM, Borok Z: Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc 2006;3(4):377-382. https://doi.org/10.1513/pats.200601-004TK
- Wynn TA: Cellular and molecular mechanisms of fibrosis. J Pathol 2008;214(2):199-210 https://doi.org/10.1002/path.2277
- van Meeteren LA, ten Dijke P: Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res 2012;347(1):177-186. https://doi.org/10.1007/s00441-011-1222-6
- Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, Ferrarini L, Orsenigo F, Papa E, Boulday G, Tournier-Lasserve E, Chapon F, Richichi C, Retta SF, Lampugnani MG, Dejana E: EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 2013;498(7455):492-496. https://doi.org/10.1038/nature12207
- Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, Tellides G, Schwartz MA, Simons M: Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest 2015;125(12):4514-4528. https://doi.org/10.1172/JCI82719
- Smeda M, Kieronska A, Adamski MG, Proniewski B, Sternak M, Mohaissen T, Przyborowski K, Derszniak K, Kaczor D, Stojak M, Buczek E, Jasztal A, Wietrzyk J, Chlopicki S: Nitric oxide deficiency and endothelial-mesenchymal transition of pulmonary endothelium in the progression of 4T1 metastatic breast cancer in mice. Breast Cancer Res 2018;20(1):86. https://doi.org/10.1186/s13058-018-1013-z
- Liu Q, Yu M, Lin Z, Wu L, Xia P, Zhu M, Huang B, Wu W, Zhang R, Li K, Zhu L, Wang Q: COL1A1-positive endothelial cells promote gastric cancer progression via the ANGPTL4-SDC4 axis driven by endothelial-to-mesenchymal transition. Cancer Lett 2025;623:217731. https://doi.org/10.1016/j.canlet.2025.217731
- Connell ND, Rheinwald JG: Regulation of the cytoskeleton in mesothelial cells: reversible loss of keratin and increase in vimentin during rapid growth in culture. Cell 1983 Aug;34(1):245-253. https://doi.org/10.1016/0092-8674(83)90155-1
- Yang WS, Kim BS, Lee SK, Park JS, Kim SB: Interleukin-1beta stimulates the production of extracellular matrix in cultured human peritoneal mesothelial cells. Perit Dial Int. 1999 May-Jun;19(3):211-220 PMID: 10433157.
- Fang CC, Yen CJ, Chen YM, Shyu RS, Tsai TJ, Lee PH, Hsieh BS: Pentoxifylline inhibits human peritoneal mesothelial cell growth and collagen synthesis: effects on TGF-beta. Kidney Int 2000;57(6):2626-2633. https://doi.org/10.1046/j.1523-1755.2000.00123.x
- Ha H, Yu MR, Lee HB: High glucose-induced PKC activation mediates TGF-beta 1 and fibronectin synthesis by peritoneal mesothelial cells. Kidney Int 2001;59(2):463-470. https://doi.org/10.1046/j.1523-1755.2001.059002463.x
- Medcalf JF, Walls J, Pawluczyk IZ, Harris KP: Effects of glucose dialysate on extracellular matrix production by human peritoneal mesothelial cells (HPMC): the role of TGF-beta. Nephrol Dial Transplant 2001;16(9):1885-1892. https://doi.org/10.1093/ndt/16.9.1885
- Yáñez-Mó M, Lara-Pezzi E, Selgas R, Ramírez-Huesca M, Domínguez-Jiménez C, Jiménez-Heffernan JA, Aguilera A, Sánchez-Tomero JA, Bajo MA, Alvarez V, Castro MA, del Peso G, Cirujeda A, Gamallo C, Sánchez-Madrid F, López-Cabrera M: Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N Engl J Med 2003;348(5):403-413. https://doi.org/10.1056/NEJMoa020809
- Sandoval P, Jiménez-Heffernan JA, Rynne-Vidal Á, Pérez-Lozano ML, Gilsanz Á, Ruiz-Carpio V, Reyes R, García-Bordas J, Stamatakis K, Dotor J, Majano PL, Fresno M, Cabañas C, López-Cabrera M. Carcinoma-associated fibroblasts derive from mesothelial cells via mesothelial-to-mesenchymal transition in peritoneal metastasis. J Pathol 2013;231(4):517-531. https://doi.org/10.1002/path.4281
- Sandoval P, Loureiro J, González-Mateo G, Pérez-Lozano ML, Maldonado-Rodríguez A, Sánchez-Tomero JA, Mendoza L, Santamaría B, Ortiz A, Ruíz-Ortega M, Selgas R, Martín P, Sánchez-Madrid F, Aguilera A, López-Cabrera M. PPAR-γ agonist rosiglitazone protects peritoneal membrane from dialysis fluid-induced damage. Lab Invest 2010;90(10):1517-1532. https://doi.org/10.1038/labinvest.2010.111
- Loureiro J, Aguilera A, Selgas R, Sandoval P, Albar-Vizcaíno P, Pérez-Lozano ML, Ruiz-Carpio V, Majano PL, Lamas S, Rodríguez-Pascual F, Borras-Cuesta F, Dotor J, López-Cabrera M. Blocking TGF-β1 protects the peritoneal membrane from dialysate-induced damage. J Am Soc Nephrol 2011;22(9):1682-1695. https://doi.org/10.1681/ASN.2010111197
- Kasashima H, Yashiro M, Kinoshita H, Fukuoka T, Morisaki T, Masuda G, Sakurai K, Kubo N, Ohira M, Hirakawa K. Lysyl oxidase is associated with the epithelial-mesenchymal transition of gastric cancer cells in hypoxia. Gastric Cancer 2016;19(2):431-442. https://doi.org/10.1007/s10120-015-0510-3
- Dellavalle A, Maroli G, Covarello D, Azzoni E, Innocenzi A, Perani L, Antonini S, Sambasivan R, Brunelli S, Tajbakhsh S, Cossu G: Pericytes resident in postnatal skeletal muscle differentiate into muscle fibres and generate satellite cells. Nat Commun 2011;2:499. https://doi.org/10.1038/ncomms1508
- Feng F, Feng X, Zhang D, Li Q, Yao L: Matrix Stiffness Induces Pericyte-Fibroblast Transition Through YAP Activation. Front Pharmacol 2021;12:698275. https://doi.org/10.3389/fphar.2021.698275
- Demuytere J, Ceelen W, Van Dorpe J, Hoorens A: The role of the peritoneal microenvironment in the pathogenesis of colorectal peritoneal carcinomatosis. Exp Mol Pathol 2020;115:104442. https://doi.org/10.1016/j.yexmp.2020.104442
- Bhattacharjee S, Hamberger F, Ravichandra A, Miller M, Nair A, Affo S, Filliol A, Chin L, Savage TM, Yin D, Wirsik NM, Mehal A, Arpaia N, Seki E, Mack M, Zhu D, Sims PA, Kalluri R, Stanger BZ, Olive KP, Schmidt T, Wells RG, Mederacke I, Schwabe RF: Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J Clin Invest 2021;131(11):e146987. https://doi.org/10.1172/JCI146987
- Liu Y, Chen S, Wan X, Wang R, Luo H, Chang C, Dai P, Gan Y, Guo Y, Hou Y, Sun Y, Teng Y, Cui X, Liu M: Tryptophan 2, 3-dioxygenase-positive matrix fibroblasts fuel breast cancer lung metastasis via kynurenine-mediated ferroptosis resistance of metastatic cells and T cell dysfunction. Cancer Commun (Lond) 2024;44(11):1261-1286. https://doi.org/10.1002/cac2.12608
- Cedric KK, Sun S, Jin D, Bao S, Sun B, Zhou J, Liu T, Kong J. LOX From Salivary Adenoid Cystic Carcinoma-Associated Fibroblast Promotes Fibrosis in the Pulmonary Pre-metastatic Niche. Int Dent J. 2026;76(1):109298. https://doi.org/10.1016/j.identj.2025.109298
- Abeysundara N, Rasnitsyn A, Fong V, Bahcheli A, Van Ommeren R, Juraschka K, Vladoiu M, Ong W, Livingston B, de Antonellis P, Ly M, Holgado BL, Sirbu O, Bahrampour S, Min HK, Fan J, Nor C, Visvanathan A, Zhang J, Wang H, Qin L, Huang N, Pallotta J, Douglas T, Mak E, Su H, Ng K, Zhang KY, Daniels C, Lucas CG, Eberhart CG, Liu H, Jiang T, Notta F, Ramaswamy V, Reimand J, Gallo M, Rich JN, Wu X, Huang X, Taylor MD: Metastatic medulloblastoma remodels the local leptomeningeal microenvironment to promote further metastatic colonization and growth. Nat Cell Biol 2025;27(5):863-874. https://doi.org/10.1038/s41556-025-01660-7
- Ramos C, Walterskirchen N, Knöbl V, Zotter C, Müller C, Gerakopoulos V, Rauch A, Falk L, Sachet M, D'Angelo E, Agostini M, Pils D, Aust S, Grusch M, Herzog R, Kratochwill K, Le Blanc S, Lenos KJ, Vermeulen L, Riss S, Bachleitner-Hofmann T, Strobel O, Dolznig H, Bergmann M, Brostjan C, Unger LW, Oehler R. Colorectal cancer peritoneal metastasis is promoted by tissue-specific fibroblasts that can arise in response to various local disorders. Cancer Lett 2025:217686. https://doi.org/10.1016/j.canlet.2025.217686
- Chung B, Esmaeili AA, Gopalakrishna-Pillai S, Murad JP, Andersen ES, Kumar Reddy N, Srinivasan G, Armstrong B, Chu C, Kim Y, Tong T, Waisman J, Yim JH, Badie B, Lee PP. Human brain metastatic stroma attracts breast cancer cells via chemokines CXCL16 and CXCL12 NPJ Breast Cancer. 2017 Mar 2;3:6. https://doi.org/10.1038/s41523-017-0008-8
- Caligiuri G, Tuveson DA: Activated fibroblasts in cancer: Perspectives and challenges. Cancer Cell 2023;41(3):434-449. https://doi.org/10.1016/j.ccell.2023.02.015
- Walterskirchen N, Müller C, Ramos C, Zeindl S, Stang S, Herzog D, Sachet M, Schimek V, Unger L, Gerakopoulos V, Hengstschläger M, Bachleitner-Hofmann T, Bergmann M, Dolznig H, Oehler R: Metastatic colorectal carcinoma-associated fibroblasts have immunosuppressive properties related to increased IGFBP2 expression. Cancer Lett 2022;540:215737. https://doi.org/10.1016/j.canlet.2022.215737
- Gui Y, Aguilar-Mahecha A, Krzemien U, Hosein A, Buchanan M, Lafleur J, Pollak M, Ferrario C, Basik M: Metastatic Breast Carcinoma-Associated Fibroblasts Have Enhanced Protumorigenic Properties Related to Increased IGF2 Expression. Clin Cancer Res 2019;25(23):7229-7242. https://doi.org/10.1158/1078-0432.CCR-19-1268
- Sansone P, Berishaj M, Rajasekhar VK, Ceccarelli C, Chang Q, Strillacci A, Savini C, Shapiro L, Bowman RL, Mastroleo C, De Carolis S, Daly L, Benito-Martin A, Perna F, Fabbri N, Healey JH, Spisni E, Cricca M, Lyden D, Bonafé M, Bromberg J: Evolution of Cancer Stem-like Cells in Endocrine-Resistant Metastatic Breast Cancers Is Mediated by Stromal Microvesicles. Cancer Res 2017;77(8):1927-1941. https://doi.org/10.1158/0008-5472.CAN-16-2129
- Fang T, Lv H, Lv G, Li T, Wang C, Han Q, Yu L, Su B, Guo L, Huang S, Cao D, Tang L, Tang S, Wu M, Yang W, Wang H: Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat Commun 2018;9(1):191. https://doi.org/10.1038/s41467-017-02583-0
- Umakoshi M, Takahashi S, Itoh G, Kuriyama S, Sasaki Y, Yanagihara K, Yashiro M, Maeda D, Goto A, Tanaka M: Macrophage-mediated transfer of cancer-derived components to stromal cells contributes to establishment of a pro-tumor microenvironment. Oncogene 2019;38(12):2162-2176. https://doi.org/10.1038/s41388-018-0564-x
- Ooi LP, Crawford DH, Gotley DC, Clouston AD, Strong RW, Gobe GC, Halliday JW, Bridle KR, Ramm GA: Evidence that "myofibroblast-like" cells are the cellular source of capsular collagen in hepatocellular carcinoma. J Hepatol 1997;26(4):798-807. https://doi.org/10.1016/s0168-8278(97)80245-0
- Mueller L, Goumas FA, Affeldt M, Sandtner S, Gehling UM, Brilloff S, Walter J, Karnatz N, Lamszus K, Rogiers X, Broering DC: Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am J Pathol 2007;171(5):1608-1618. https://doi.org/10.2353/ajpath.2007.060661
- Mueller L, von Seggern L, Schumacher J, Goumas F, Wilms C, Braun F, Broering DC. TNF-alpha similarly induces IL-6 and MCP-1 in fibroblasts from colorectal liver metastases and normal liver fibroblasts. Biochem Biophys Res Commun. 2010 Jul 2;397(3):586-91. https://doi.org/10.1016/j.bbrc.2010.05.163
- O'Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, Dewar R, Rocha RM, Brentani RR, Resnick MB, Neilson EG, Zeisberg M, Kalluri R: VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci USA 2011;108(38):16002-7. https://doi.org/10.1073/pnas.1109493108
- Raz Y, Cohen N, Shani O, Bell RE, Novitskiy SV, Abramovitz L, Levy C, Milyavsky M, Leider-Trejo L, Moses HL, Grisaru D, Erez N: Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J Exp Med 2018;215(12):3075-3093. https://doi.org/10.1084/jem.20180818
- Kong J, Tian H, Zhang F, Zhang Z, Li J, Liu X, Li X, Liu J, Li X, Jin D, Yang X, Sun B, Guo T, Luo Y, Lu Y, Lin B, Liu T: Extracellular vesicles of carcinoma-associated fibroblasts creates a pre-metastatic niche in the lung through activating fibroblasts. Mol Cancer 2019;18(1):175. https://doi.org/10.1186/s12943-019-1101-4
- Cai J, Tang H, Xu L, Wang X, Yang C, Ruan S, Guo J, Hu S, Wang Z: Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness. Carcinogenesis 2012;33(1):20-29. https://doi.org/10.1093/carcin/bgr230
- Olaso E, Salado C, Egilegor E, Gutierrez V, Santisteban A, Sancho-Bru P, Friedman SL, Vidal-Vanaclocha F: Proangiogenic role of tumor-activated hepatic stellate cells in experimental melanoma metastasis. Hepatology 2003;37(3):674-685. https://doi.org/10.1053/jhep.2003.50068
- Ishii G, Sangai T, Oda T, Aoyagi Y, Hasebe T, Kanomata N, Endoh Y, Okumura C, Okuhara Y, Magae J, Emura M, Ochiya T, Ochiai A: Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem Biophys Res Commun 2003;309(1):232-240. https://doi.org/10.1016/s0006-291x(03)01544-4
- Chauhan H, Abraham A, Phillips JR, Pringle JH, Walker RA, Jones JL: There is more than one kind of myofibroblast: analysis of CD34 expression in benign, in situ, and invasive breast lesions. J Clin Pathol 2003;56(4):271-6. https://doi.org/10.1136/jcp.56.4.271
- Rynne-Vidal A, Au-Yeung CL, Jiménez-Heffernan JA, Pérez-Lozano ML, Cremades-Jimeno L, Bárcena C, Cristóbal-García I, Fernández-Chacón C, Yeung TL, Mok SC, Sandoval P, López-Cabrera M: Mesothelial-to-mesenchymal transition as a possible therapeutic target in peritoneal metastasis of ovarian cancer. J Pathol 2017;242(2):140-151. https://doi.org/10.1002/path.4889
- Krizbai IA, Gasparics Á, Nagyőszi P, Fazakas C, Molnár J, Wilhelm I, Bencs R, Rosivall L, Sebe A: Endothelial-mesenchymal transition of brain endothelial cells: possible role during metastatic extravasation. PLoS One 2015;10(3):e0123845. https://doi.org/10.1371/journal.pone.0123845
- Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, Friedman R, Varro A, Tycko B, Wang TC: Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011;19(2):257-272. https://doi.org/10.1016/j.ccr.2011.01.020
- Bochet L, Lehuédé C, Dauvillier S, Wang YY, Dirat B, Laurent V, Dray C, Guiet R, Maridonneau-Parini I, Le Gonidec S, Couderc B, Escourrou G, Valet P, Muller C: Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res 2013;73(18):5657-68. https://doi.org/10.1158/0008-5472.CAN-13-0530
- Hosaka K, Yang Y, Seki T, Fischer C, Dubey O, Fredlund E, Hartman J, Religa P, Morikawa H, Ishii Y, Sasahara M, Larsson O, Cossu G, Cao R, Lim S, Cao Y: Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc Natl Acad Sci USA 2016;113(38):E5618-27. https://doi.org/10.1073/pnas.1608384113
- Luo H, Xia X, Huang LB, An H, Cao M, Kim GD, Chen HN, Zhang WH, Shu Y, Kong X, Ren Z, Li PH, Liu Y, Tang H, Sun R, Li C, Bai B, Jia W, Liu Y, Zhang W, Yang L, Peng Y, Dai L, Hu H, Jiang Y, Hu Y, Zhu J, Jiang H, Li Z, Caulin C, Park J, Xu H: Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat Commun 2022;13(1):6619. https://doi.org/10.1038/s41467-022-34395-2
- Saini P, Mirji G, Islam SMS, Simons LM, Bhat SA, Bonfanti AP, Muthumani K, Agrawal P, Cassel J, Tang HY, Tateno H, Zhang R, Hultquist JF, Shinde RS, Abdel-Mohsen M: Targeting Interactions Between Siglec-10 and α3β1 Integrin Enhances Macrophage-Mediated Phagocytosis of Pancreatic Cancer. Cancer Res 2026;86(1):99-115. https://doi.org/10.1158/0008-5472.CAN-25-0977
- Munday J, Kerr S, Ni J, Cornish AL, Zhang JQ, Nicoll G, Floyd H, Mattei MG, Moore P, Liu D, Crocker PR: Identification, characterization and leucocyte expression of Siglec-10, a novel human sialic acid-binding receptor. Biochem J 2001;355(Pt 2):489-497. https://doi.org/10.1042/0264-6021:3550489
- Li N, Zhang W, Wan T, Zhang J, Chen T, Yu Y, Wang J, Cao X: Cloning and characterization of Siglec-10, a novel sialic acid binding member of the Ig superfamily, from human dendritic cells. J Biol Chem 2001;276(30):28106-28112. https://doi.org/10.1074/jbc.M100467200
- Chen GY, Tang J, Zheng P, Liu Y: CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science 2009;323(5922):1722-1725. https://doi.org/10.1126/science.1168988
- Kristiansen G, Winzer KJ, Mayordomo E, Bellach J, Schlüns K, Denkert C, Dahl E, Pilarsky C, Altevogt P, Guski H, Dietel M: CD24 expression is a new prognostic marker in breast cancer. Clin Cancer Res 2003;9(13):4906-4613 PMID: 14581365.
- Zheng J, Li Y, Yang J, Liu Q, Shi M, Zhang R, Shi H, Ren Q, Ma J, Guo H, Tao Y, Xue Y, Jiang N, Yao L, Liu W: NDRG2 inhibits hepatocellular carcinoma adhesion, migration and invasion by regulating CD24 expression. BMC Cancer 2011;11:251:1-9. https://doi.org/10.1186/1471-2407-11-251
- Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, Krishnan V, Hatakeyama J, Dorigo O, Barkal LJ, Weissman IL: CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019;572(7769):392-396. https://doi.org/10.1038/s41586-019-1456-0
- Zhang P, Lu X, Tao K, Shi L, Li W, Wang G, Wu K: Siglec-10 is associated with survival and natural killer cell dysfunction in hepatocellular carcinoma. J Surg Res 2015;194(1):107-113. https://doi.org/10.1016/j.jss.2014.09.035
- Xiao N, Zhu X, Li K, Chen Y, Liu X, Xu B, Lei M, Xu J, Sun HC: Blocking siglec-10hi tumor-associated macrophages improves anti-tumor immunity and enhances immunotherapy for hepatocellular carcinoma. Exp Hematol Oncol 2021;10(1):36. https://doi.org/10.1186/s40164-021-00230-5
- Canè S, Bronte V: The sialic shield of leukemia cells. Science 2026;392(6794):147-148. https://doi.org/10.1126/science.aeg6715
- Lipponen P, Aaltomaa S, Kosma VM, Syrjänen K: Apoptosis in breast cancer as related to histopathological characteristics and prognosis. Eur J Cancer 1994;30A(14):2068-2073. https://doi.org/10.1016/0959-8049(94)00342-3
- Carrel A: Mechanism of the formation and growth of malignant tumors. Ann Surg 1925;82(1):1-13. https://doi.org/10.1097/00000658-192507010-00001
- Fischer A: The theory of the developmental physiology of malignant tumors. Am J Cancer 1937;31:1–20. https://doi.org/10.1158/ajc.1937.1b
- Wang RA, Li QL, Li ZS, Zheng PJ, Zhang HZ, Huang XF, Chi SM, Yang AG, Cui R: Apoptosis drives cancer cells proliferate and metastasize. J Cell Mol Med 2013;17(1):205-211. https://doi.org/10.1111/j.1582-4934.2012.01663.x
- Rous P: A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J Exp Med 1911;13(4):397-411. https://doi.org/10.1084/jem.13.4.397
- Rous P: Surmise and fact on the nature of cancer. Nature 1959;183(4672):1357-1361. https://doi.org/10.1038/1831357a0
- Rous P: The challenge to man of the neoplastic cell. Science. 1967 Jul 7;157(3784):24-28. https://doi.org/10.1126/science.157.3784.24
- Weiss RA, Vogt PK: 100 years of Rous sarcoma virus. J Exp Med 2011;208(12):2351-2355. https://doi.org/10.1084/jem.20112160
- Vrantsidis TH, Lombrozo T: Inside Ockham's razor: A mechanism driving preferences for simpler explanations. Mem Cognit 2025;53(3):746-774. https://doi.org/10.3758/s13421-024-01604-w
- Panchy N, Watanabe K, Takahashi M, Willems A, Hong T: Comparative single-cell transcriptomes of dose and time dependent epithelial-mesenchymal spectrums. NAR Genom Bioinform 2022;4(3):lqac072. https://doi.org/10.1093/nargab/lqac072
- Ruscetti M, Quach B, Dadashian EL, Mulholland DJ, Wu H: Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res 2015;75(13):2749-2759. https://doi.org/10.1158/0008-5472.CAN-14-3476
- Buissant des Amorie JR, Hageman JH, Brunner SR, van der Horst SEM, Puschhof MC, van Hoeck A, van Lierop I, Middelkamp S, van der Schee L, van Kempen S, Morsink F, Geene R, Mertens S, Cavigelli DS, Verlaan-Klink I, Kraaier LJ, Salij J, Bezemer R, Kranenburg O, Laclé MM, Moons LMG, Snippert HJG: Emergence of oncofetal plasticity is ubiquitous in early colorectal cancers. Nature 2026 Apr 15. Online ahead of print. https://doi.org/10.1038/s41586-026-10344-7
- Chen X, Winter M, Rokavec M, König J, Hermeking H: TGF-β/SMAD4/14-3-3σ/TFEB axis promotes mesenchymal-epithelial transition and inhibits autophagy in colorectal cancer. Cell Death Dis 2026;17(1):397. https://doi.org/10.1038/s41419-026-08733-x
- Sasaki A, Hosonuma M, Maruyama Y, Funayama E, Toyoda H, Tsurui T, Tajima K, Nakashima R, Yamazaki Y, Orimo A, Oguchi T, Yoshimura K, Kuramasu A: Cetirizine suppresses cancer-associated fibroblast-mediated fibrotic remodeling and epithelial-mesenchymal transition via histamine H1 receptor inverse agonism. Breast Cancer Res 2026 Apr 26 Online ahead of print. https://doi.org/10.1186/s13058-026-02284-x
- Zhao D, Xi Y, Shao J, Ding X, Wang W, Jin Y, Li Z, Li Z, Zhang X: Photodynamic therapy remodels the prostate cancer microenvironment by suppressing cancer-associated fibroblast-mediated calcium signaling. J Photochem Photobiol B 2026;279:113440. Online ahead of print. https://doi.org/10.1016/j.jphotobiol.2026.113440
- Fidler IJ, Cifone MA: Properties of metastatic and nonmetastatic cloned subpopulations of an ultraviolet-light-induced murine fibrosarcoma of recent origin. Am J Pathol 1979;97(3):633-648. PMID: 507195.
- Mencken HL: The Divining Afflatus. New York Evening Mail. Nov 16, 1917.
- Mencken HL: Prejudices: Second Series. Jonathan Cape, 11 Gower Street, London. Page 158 1921
- Billroth T: Brief Theodor Billroth an Prof. Baum in Göttingen. 09. August 1877. In: Briefe von Theodor Billroth. Von Dr. Georg Fischer. Erste Auflage. Georg Fischer Verlag. 1896.