Original Article – DOI: 10.33594/000000876
CPB (60): 346 - 375
Accepted: 01.07.2026 - Published: 14.07.2026
Chronic Infection and Cardiac Aging: a New Perspective on Pathogen-Associated Cardiomyopathy
bBinghamton Biofilms Research Center, Binghamton University, Binghamton, NY, USA,
cDepartment of Pharmaceutical Sciences, Binghamton University School of Pharmacy and Pharmaceutical Sciences, Binghamton, NY, USA
Keywords
Abstract
Cardiovascular diseases remain the primary driver of global mortality, with advanced age serving the most significant risk factor for their development and progression. Emerging evidence suggests that chronic infections can act as potent catalysts for cardiac decline by prematurely inducing aging phenotypes. Pathogens, including viruses, bacteria, and parasites, that evade host clearance establish a state of permanent inflammaging: a chronic, low-grade inflammatory milieu characterized by persistent cytokine signaling and leukocyte infiltration. This environment directly mirrors the sterile inflammation that drives natural senescence. Mechanistically, chronic infection subverts the heart’s homeostatic pathways, triggering cardiomyocyte senescence through the dysregulation of mTOR signaling and the impairment of autophagy. These infections further drive mitochondrial dysfunction and the overproduction of reactive oxygen species (ROS), leading to oxidative DNA damage and metabolic exhaustion within the myocardium. On a structural level, immune subversion, via macrophage polarization and the induction of autoimmunity, accelerates left ventricular hypertrophy, myocardial remodeling, and interstitial fibrosis. By characterizing chronic infection as a modifiable driver of biological aging, we can prioritize anti-infective strategies as essential components of cardiovascular longevity and geriatric care.
Introduction
Heart disease is the leading cause of death in the United States, accounting for over 600, 000 deaths in 2023 (1). Cardiovascular diseases become more common with age in both men and women (2). As the heart ages, left ventricular hypertrophy, myocardial remodeling, and cardiomyocyte senescence lead to diseased states such as heart failure, arrhythmia, and atrial fibrillation (3, 4). As such, slowing or preventing age-related cardiac decline is of great interest to prolong life- and health-span for millions while reducing burden on healthcare systems. Age is increasingly understood as an inflammatory process, characterized by chronic low-grade inflammation called inflammaging, which can be influenced by infectious diseases (5, 6). Infectious agents such as viruses, parasites, and bacteria can directly damage the heart via cardiomyocyte infection and secreted virulence factors, and induce inflammation leading to secondary damage to myocardium, such as fibrosis and hypertrophy, via the immune system (7). Many infections can evade clearance by the immune system to enter a chronic state, and continuously inducing a low-grade inflammatory response accelerating cardiac damage (8). Chronic infections promote several aging-related factors, including elevated reactive oxygen species (ROS) production, mitochondrial dysfunction, impaired autophagy, dysregulated mTOR signaling, and metabolic alterations, all of which can contribute to persistent low-grade inflammation (9–13). These intertwined factors may result in a pro-aging milieu advancing cardiovascular aging. By understanding these changes, we can emphasize the importance of proper prevention and treatment of cardiotropic infections and may discover remediation for cardiac damage and senescence. Here, we review recent evidence linking chronic infectious diseases with the development and progression of cardiac aging phenotypes.
Hallmarks of Cardiovascular Aging
The aging heart undergoes pathological structural and functional changes, including left ventricular hypertrophy, myocardial remodeling, heart failure (with or without reduced left ventricular ejection fraction), arrhythmia, and atrial fibrillation (3, 4). At the molecular level, cardiac aging is driven by several interconnected mechanisms including prolonged inflammatory damage, reactive oxygen species (ROS) production and oxidative damage, mitochondrial dysfunction, and impaired autophagy causing cardiomyocyte senescence, tissue damage, fibrosis, and ultimately age-related performance declines such as heart failure (3, 14). Excessive ROS generation leads to DNA and mitochondrial damage, telomere shortening, and disrupted energy metabolism, establishing a vicious cycle of oxidative and inflammatory injury. Additionally, ATP synthesis is reduced, leading to a greater reliance on glycolysis and reduced ATP reserves. Simultaneously, defective autophagy and persistent mTOR activation exacerbate protein aggregation, fibrosis, and cellular stress responses. Chronic infections amplify these factors, accelerating age-related decline in cardiac function. Among these hallmarks, inflammation is central in driving cardiac aging. Persistent activation of inflammatory pathways contributes to the development of dilated cardiomyopathy (DCM), myocardial fibrosis, and heart failure (15–17). In response to injury or infection, inflammatory cells like macrophages, T-cells, and eosinophils infiltrate the myocardium, releasing proinflammatory cytokines and contributing to pathological cardiac remodeling (15, 17, 18). These cytokines activate nuclear factor kappa B (NF-κB), a transcription factor which induces transcription of inflammatory mediators and the NLRP3 inflammasome (19, 20). The NLRP3 inflammasome induces pyroptosis, highly inflammatory cell death which releases cell contents into the extracellular space, causing cardiac damage (21, 22). Senescent cardiomyocytes further sustain inflammation through the senescence-associated secretory phenotype (SASP), perpetuating a milieu of cytokine release, mitochondrial dysfunction, and hypertrophy (23). Thus, inflammation, often sustained by chronic infections or autoimmune responses, acts as a key driver of cardiac aging. In the following sections we discuss these changes in more details. Table 1 summarizes the key hallmarks of cardiac aging and highlights analogous pathological features observed across a range of infectious diseases.
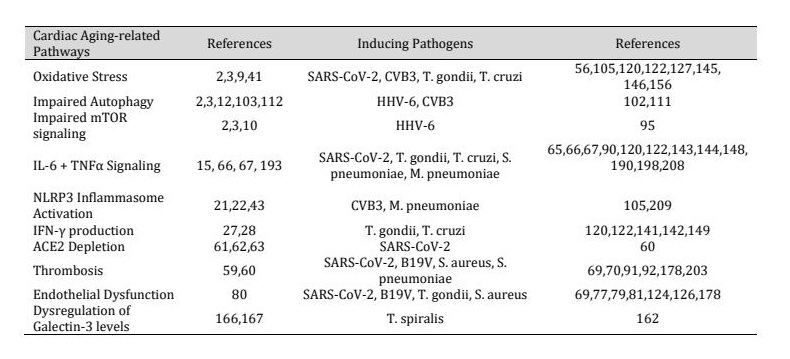
Table 1: Similarities between cardiac aging and symptoms of infectious diseases
Immune Changes in Cardiac Aging
As we age, the immune system is affected by a series of changes termed “immunosenescence” including changes to immune cell populations, increased cytokine production, and the formation of autoantibodies (24). The progression of immunosenescence and the resulting chronic inflammation, often called inflammaging, lacks a central marker. Instead, aging patients show an increase in immune signaling overall, as tracked by multiple inflammatory and anti-inflammatory markers (25). In aged mice, the accumulation of activated CD4⁺ T cells promotes chronic low-grade inflammation in the heart. This leads to increased immune cell infiltration, myocardial remodeling, fibrosis, and mild cardiac dysfunction; notably, depletion of lymphocytes or transfer of young immune cells attenuates these age-related cardiac changes (26). In a recent study, T-cells were collected from the mediastinal lymph nodes which surround the heart of young and aged mice and subjected to single-cell RNA sequencing. Analysis revealed an age-associated increase in cytotoxic CD8⁺ T-cells and effector memory CD4⁺ T cells (27). In both CD8+ and CD4+ T-cells, from the lymph nodes or myocardium, IFN-γ levels increased, as did expression of IFN-γ response genes (27). This is notable as IFN-γ treatment has been shown to increase the risk of arrythmia and atrial fibrillation in rats (28). In the same study, CD8+ T-cells expressing IFN-γ in patient peripheral and atrial blood were closely associated with atrial fibrillation and post-procedural recurrence thereof, corroborating the rat results (28). It has been suggested that T-cells activate fibroblasts leading to collagen deposition and myofibroblast differentiation (29, 30). T-cell deficient mice were transplanted with naïve, Th1 wild type, and Th1 IFN-γ knockout T-cells following thoracic aortic constriction to induce non-ischemic heart failure. Only mice with mature IFN-γ competent T-cells developed significant perivascular fibrosis, showing that IFN-γ is necessary for T-cell mediated fibrosis (31). Similarly, major histocompatibility complex II (MHCII) was shown to be necessary for age-related cardiac decline, as MHCII knockout rescued age-related decline in fractional shortening and end diastolic area in mice (32). T-cell changes alone cannot replicate an aged-like phenotype, however, as adoptive transfer of aged T-cells to young mice was insufficient to induce cardiac decline (32). Age-related changes to immune cell populations are not limited to T-cells. Macrophages undergo significant population shifts and are a source of increased inflammation with age. Cardiac macrophages are of one of two lineages: embryonic yolk sac derived macrophages which are present from birth and proliferate in situ, and CCR2+ monocyte derived macrophages which infiltrate into the myocardium from the bloodstream (17). In a study of young and aged female mice, the total number of macrophages increased with age with a greater proportion of CCR2+ monocyte derived macrophages among their number (33). Among both lineages, multiple inflammatory pathways are upregulated, leading to a more inflammatory phenotype. Many inflammatory signals, including IL-1β, IL-6, IL-18, and TNFα, have been associated with cardiovascular disease and may induce profibrotic cytokine release (29, 34). While both lineages expressed proinflammatory pathways, infiltrating CCR2+ macrophages may be more important to profibrotic pathways, as in mouse models of cryoinjury and repeated ischemic insult, inhibition of macrophage infiltration significantly reduced collagen deposition (I.E. fibrosis)(35, 36). The inflammatory signals released by infiltrating and resident immune cells, as well as molecular patterns from pathogens or cellular damage recognized by pattern recognition receptors, activate the transcription factor NF-κB which in turn promotes pro-inflammatory signaling, immune activity, and cell survival (20). In humans and mice NF-κB activity increases with age and stress which may be mediated by epigenetic regulation of FK506-binding protein 51 (FKBP51), a protein cochaperone which interacts with NF-κB regulators (37–39). To study the role of NF-κB in cardiac aging, mice were modified to overexpress myotroponin to induce cardiac hypertrophy. The mice were then treated with a viral vector for small interfering RNA against the p65 subunit of NF-κB, decreasing heart weight to body mass ratio and left atrial area and increasing ejection fraction and fractional shortening, indicating improved diastolic function (40). In a later study, male mice were treated with D-galactose to induce premature aging via oxidative stress, reducing ejection fraction, fractional shortening, and telomere length and upregulating TNF-α, all of which was prevented by exosome treatment (39). Then, in an in vitro senescence model, the same exosome treatment was shown to reduce inflammation and senescence identically to NF-κB inhibition (39). Taken together, these results suggest that NF-κB is a key regulator of inflammatory cardiac damage in aging. Among the genes upregulated by NF-κB are the components of the NLRP3 inflammasome, a large protein complex which cleaves IL-1β and IL-18 into their active forms by facilitating the cleavage and activation of procaspase-1 (19). NF-κB primes the inflammasome by upregulating its components, and then assembly is induced by endogenous signals including ROS released by damaged mitochondria often found in aging cardiomyocytes (41, 42). Expression of related inflammasomes, inflammasome related genes, and IL-1β, which signals NF-κB activation, is increased in older patients and correlates with risk for hypertension, pulse-wave velocity (a measure of arterial stiffness), and lower self-reported familial longevity (43). NLRP3 expression is necessary for cardiomyocyte senescence, structural remodeling, and reduced contractile performance from D-galactose treatment in mice hearts and in rat cardiomyocytes (21, 22). Therefore, NF-κB may mediate inflammatory cardiac aging via the NLRP3 inflammasome. The inflammasome itself is certainly involved in cardiomyocyte senescence, as NLRP3 inhibition attenuated D-galactose induced senescence of H9C2 cardiomyocytes in vitro (22). The NLRP3 inflammasome induces pyroptosis which releases cell contents causing severe inflammation which may contribute to further inflammatory damage (19). Fig. 1 summarizes immune alterations occur in cardiac aging.
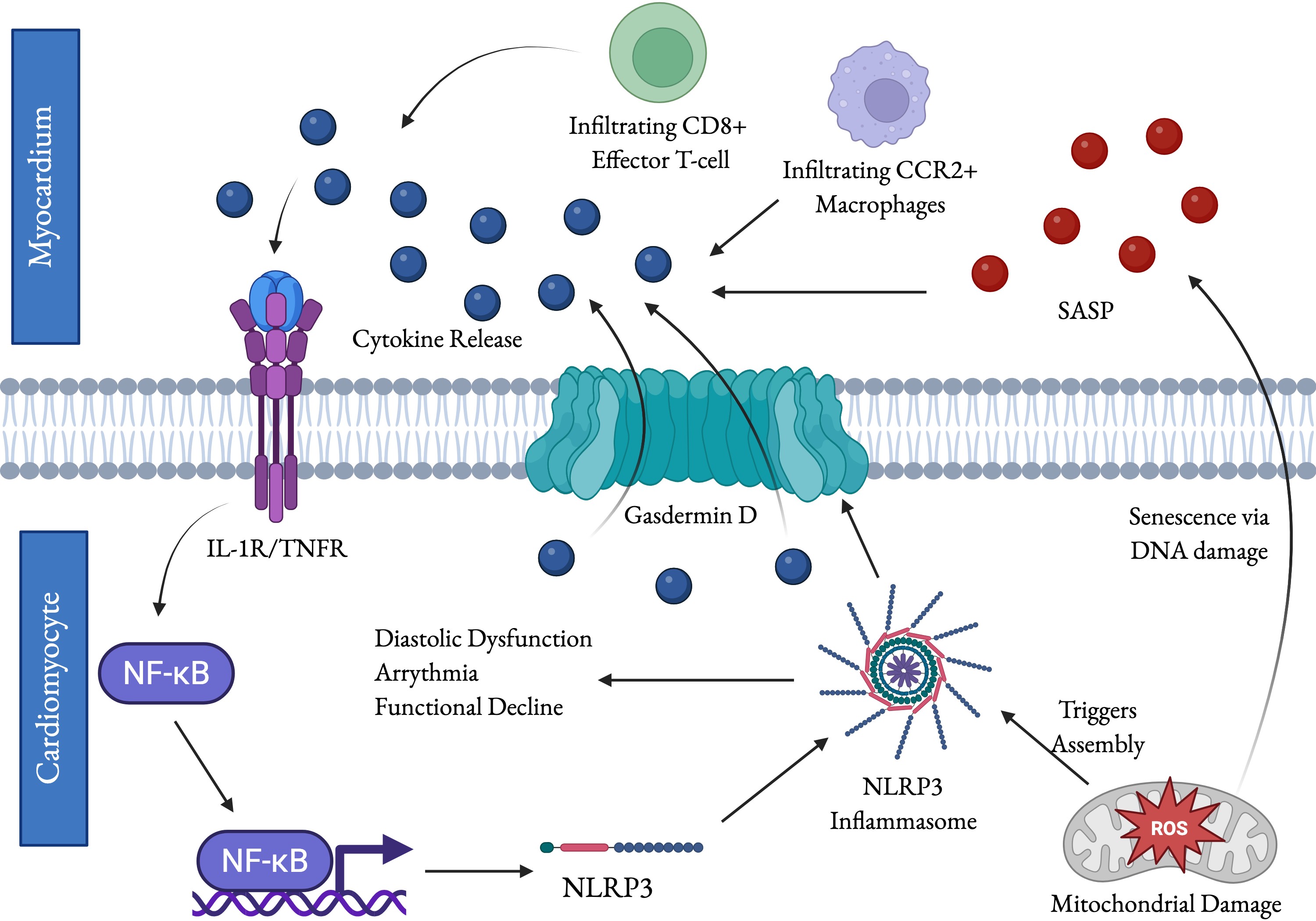
Fig. 1: Immunological alterations contribute to age-related cardiac functional decline. Aging is marked by an increase in cytokine production and a change in T-cell and macrophage populations. The increased release of inflammatory cytokines activates NF-κB, which activates transcription of inflammatory mediator genes including the NRLP3 inflammasome. Assembly of the components into the active inflammasome can be triggered by endogenous signals like ROS from mitochondrial damage. ROS also induces senescence in cardiomyocytes via DNA damage, which further contributes to cytokine release via the senescence-associate secretory phenotype (SASP). The NLRP3 inflammasome triggers pyroptosis, which releases proinflammatory factors through the formation of gasdermin D pores and is highly pro-inflammatory. NLRP3, NF-κB, certain cytokines, mitochondrial dysfunction, ROS, and immunosenescence are directly related to age-related functional decline in the heart. Fig. created by BioRender.
The Use of Artificial Intelligence-ECG in Predicting Heart Biological Age
Cardiac aging is characterized by the progressive deterioration of cardiac structure and function and is driven by cumulative molecular, cellular, and metabolic alterations (44). Hallmarks of the aging heart include increased myocardial stiffness, impaired diastolic function, mitochondrial dysfunction, chronic low-grade inflammation, and diminished capacity to respond to physiological stress, all of which contribute to an increased risk of cardiovascular disease and heart failure (44, 45). Although chronological age is a major determinant of cardiovascular risk, it does not fully capture interindividual differences in the biological processes that govern cardiac aging (46). To address this limitation, deep learning algorithms trained on large-scale datasets of standard 12-lead electrocardiograms (ECGs) have been developed to estimate biological heart age. Recent advances in artificial intelligence (AI) enable estimation of biological age using ECGs. Unlike conventional ECG interpretation, which relies on overt cardiac abnormalities, AI models capture complex latent waveform features associated with diffuse myocardial remodeling, extracellular matrix deposition, ion channel perturbations, altered repolarization dynamics, and loss of autonomic balance. As a result, AI-ECG can detect subclinical cardiovascular deterioration before the appearance of overt structural disease or measurable declines in cardiac function (47). When biological age, based on AI-ECG, exceeds chronological age by approximately 6 years, all-cause mortality and cardiovascular event risk increases (48) and age gaps greater than 8 years are associated with significantly higher mortality rate (49), highlighting the utility of AI-derived heart age as a sensitive biomarker of cardiovascular health and aging. Another clinical study evaluated 25,144 adults without pre-existing coronary artery disease, stroke, or atrial fibrillation and followed them for an average of 12.4 years. Compared with individuals whose AI-ECG age was within one standard deviation (SD) of their chronological age, those with an Age-Gap ≥1 SD exhibited significantly higher all-cause and cardiovascular disease (CVD) mortality, whereas individuals with an Age-Gap ≤−1 SD experienced lower all-cause and CVD mortality. Importantly, these associations remained significant after adjustment for traditional cardiovascular risk factors, demonstrating that the AI-ECG age gap is an independent biomarker of biological aging and a powerful predictor of long-term cardiovascular outcomes (50). Importantly, cardiovascular aging is not a static process but rather a dynamic trajectory that evolves over time. A recent study demonstrated that longitudinal assessment of multiple AI-ECGs substantially improves survival prediction compared with a single ECG measurement. In this cohort of 46,960 patients encompassing 337,415 ECGs and a median follow-up of 4.5 years, it was found that patients whose biological age exceeded their chronological age had a higher hazard of death. Notably, models incorporating serial ECG measurements significantly excelled single-ECG models in predicting all-cause mortality (51). These findings demonstrate that the AI-derived age gap is a dynamic biomarker that captures ongoing physiological changes in cardiovascular health, providing a sensitive measure of age-related decline and a potential tool for monitoring responses to interventions aimed at slowing or reversing cardiac aging. In chronic infections, a positive age gap may reflect accelerated cardiac senescence. Chronic infections create a sustained microenvironment that accelerates the heart's biological clock through four converging mechanisms. First, unlike acute infections that resolve, chronic infections sustain elevated pro-inflammatory cytokines over months to years. This chronic inflammaging directly damages cardiomyocytes, promotes interstitial fibrosis, and impairs mitochondrial function, all hallmarks of biological cardiac aging that would normally only emerge in older individuals. Second, persistent pathogen burden forces the immune system into a state of exhaustion and metabolic reprogramming (52, 53). This can cause systemic insulin resistance, oxidative stress, and mitochondrial inefficiency that mirror the metabolic profile of an aging heart, even in young patients. The infected heart is forced to operate in a chronically energy-deprived state, accelerating functional decline. Third, repeated cycles of subclinical injury and repair deposit collagen and stiffen the myocardium. This structural remodeling is indistinguishable, at the tissue level, from age-related fibrosis. It alters electrical conduction, increases arrhythmic risk, and reduces diastolic compliance, all features that AI-ECG systems detect as signatures of an older heart. Lastly, chronic infections can impair the autonomic nervous system, reducing heart rate variability and blunting baroreflex sensitivity. Autonomic aging is one of the strongest predictors of cardiovascular mortality, and chronic infection accelerates it substantially. Because gap age can be assessed non-invasively and repeatedly over time, AI-ECG offers a dynamic biomarker for monitoring disease progression and evaluating the effectiveness of interventions targeting aging-associated pathways.
Chronic Infections and Cardiovascular Aging
Viral infections
Viral infections are the major cause of myocarditis and can cause debilitating cardiac tissue damage. The infection can lead to ROS generation, chronic inflammation, and fibrosis. Infection with cardiotropic viruses such as SARS-CoV-2, Human Parvovirus B19, Human Herpes-Virus Type 6, and Coxsackievirus B lead to cardiac functional decline and molecular changes that resemble age-related decline.
SARS-CoV2. COVID-19, caused by SARS-CoV-2, demonstrates a complex pathology that not only causes acute cardiac injury, but also appears to accelerate cardiovascular aging through mechanisms of chronic inflammation, mitochondrial dysfunction, and structural damage (54–57). The heart highly expresses ACE2 (angiotensin-converting enzyme 2), the viral entry receptor of SARS-CoV-2 (58, 59). During SARS-CoV-2 infection, ACE2 dysregulation emerges as a central driver of cardiac dysfunction. As the virus binds to ACE2, it induces its downregulation or cleavage (60), disrupting one of the heart’s primary mechanisms for regulating stress responses and vascular tone (61). This feature closely mirrors the physiology of the aging heart, where the gradual age-related loss of ACE2 reduces cardiac resilience and leaves the myocardium increasingly vulnerable to injury and dysfunction (62, 63). The post-acute sequelae of COVID-19, commonly known as Long COVID, appear to resemble a state of accelerated biological aging (64), primarily through persistent inflammation and immune dysregulation (65). This chronic inflammatory state mirrors inflammaging, characterized by sustained elevations of pro-inflammatory cytokines, particularly IL-6 and TNF-α which are key mediators of age-related cardiovascular disease such as myocardial infarction and heart failure (66, 67). Persistent autoimmunity, exemplified by the presence of antiphospholipid antibodies in COVID-19 patients (68), contributes to vascular injury, thrombosis, and endothelial dysfunction (69), thereby promoting premature vascular aging. Moreover, viral spike protein binds to fibrin and fibrinogen, two central components of the coagulation pathway, which can lead to abnormal protein folding and clot formation in patient blood samples (70). Importantly, serum samples from COVID-19 patients with asymptomatic, mild, and severe disease were found to contain elevated levels of fibrin-targeting autoantibodies (70). potentially influenced by the persistence of spike protein post infection in certain cell types such as monocytes (71). Mitochondrial dysfunction, a defining hallmark of cellular aging, is increasingly recognized as a central feature of COVID-19 pathology. The virus disrupts mitochondrial structure and function across multiple cell types, leading to loss of membrane potential, excessive ROS generation, and impaired ATP production in monkey kidney and human hepatocarcinoma cells (72). In human stem cell-derived cardiomyocytes, infection reduces mitochondrial number and causes visible swelling, resulting in the cessation of rhythmic beating (73). These changes were associated with induction of cell cycle arrest in infected cardiomyocytes. Moreover, transcriptional profiling of infected cardiomyocytes further reveals a metabolic reprogramming, characterized by the downregulation of oxidative phosphorylation (74). This metabolic dysregulation is reminiscent of the metabolic decline seen in aged cardiomyocytes (75, 76). Compounding this dysfunction, SARS-CoV-2 also inhibits mitophagy, the process responsible for clearing damaged mitochondria, in monkey kidney and human hepatocarcinoma cells (72). The resulting accumulation of defective mitochondria amplifies oxidative stress and accelerates cellular senescence, driving long-term consequences such as reduced global longitudinal strain and diastolic impairment, cardiac features that closely mirror aging (77). Collectively, these mechanisms suggest that SARS-CoV-2 infections initiate and maintain processes indistinguishable from those driving natural cardiovascular aging, effectively accelerating decline in the heart and vasculature.
Human Parvovirus B19. Human Parvovirus B19 (B19V) is increasingly implicated in long-term cardiovascular pathology. While it primarily infects endothelial and erythroid progenitor cells (8), its mechanisms of damage, endothelial dysfunction with impaired regeneration and autoimmunity, significantly augment the biological processes of cardiac aging. B19V directly targets the vascular system, inducing damage and disrupting the repair processes essential for maintaining normal cardiac function. Although B19V primarily replicates in erythroid cells, it can also infect endothelial cells where viral transcription and protein synthesis lead to endothelial dysfunction and apoptosis (78, 79), a core feature of vascular aging that contributes to arterial stiffness and impaired blood flow (80). The infection further impairs endothelial regeneration by damaging endothelial progenitor cells, impairing vascular repair and angiogenesis in a mouse transplantation model (81). In addition, B19V infects bone marrow-derived circulating angiogenic cells that normally contribute to blood vessel development and repair. By disrupting these cell populations, B19V not only hinders proper vascular regeneration, but also creates conditions that facilitate its spread, ultimately promoting the progression of cardiomyopathy in B19V patients (81). Infected endothelial cells also upregulate Tenascin C and transforming growth factor beta (TGF-β) in human micro-endothelial cells (82), both of which promote cardiac fibrosis and pathological tissue remodeling (83, 84), key structural hallmarks of the aging heart. Moreover, the B19V capsid protein, VP1u, activates stress signaling pathways involving JNK, ERK, c-Jun, and c-Fos in primary rat endothelial cells (85). These molecular cascades are linked to cardiac remodeling and vascular aging that heighten long-term cardiovascular risk and mortality (86). Together, these mechanisms position B19V as a potent inducer of endothelial senescence and cardiac aging. Chronic B19V infection may compromise the heart’s energy metabolism, a process closely linked to cardiac aging. Transcriptionally active B19V, linked to higher mortality in non-ischemic cardiomyopathy patients (87), directly disrupts the expression of genes that encode essential mitochondrial respiratory chain proteins in endomyocardial biopsies (88). Such mitochondrial dysfunction not only weakens the myocardium’s ability to tolerate physiological stress but also drives the progressive decline in cardiac performance characteristic of age-related heart disease (76). B19V infection may induce a persistent autoimmune response that imitates and amplifies inflammaging. B19V antigens resemble host proteins leading to the generation of cross-reactive antibodies (44). This cross-reactivity can provoke the synthesis of autoantibodies that attack cardiac proteins, such as myosin and troponin, in B19V patients (89). Antibodies directed against the viral VP1u region, for instance, drive robust inflammation through elevated IL-6, TNF-α, and NF-κB signaling while promoting complement-mediated cardiomyocyte lysis in mice (90). B19V induced autoimmunity resembles antiphospholipid syndrome. This process is characterized by generation of antibodies against the mitochondrial phospholipid cardiolipin and the clotting regulator beta-2-glycoprotein (91, 92). This manifests as excessive clotting, vascular inflammation, coagulation abnormalities, heightened risk of stroke, valvular injury, and coronary artery disease (63, 64), all of which can lead to vascular aging and chronic inflammation.
Human Herpes-virus Type 6. Human Herpesvirus-6 (HHV-6) may accelerate cardiac aging through its ability to establish a persistent infection in the heart (8). HHV-6 infection is linked to myocarditis, DCM, and heart failure in a significant portion of infected patients as detected by endomyocardial biopsy (93). HHV-6 directly targets fundamental biological drivers of senescence, such as mitochondrial dysfunction and inflammation. HHV-6 infection induces mitochondrial alterations including increased fragmentation, and alteration of mitochondrial proteins involved in fatty acid, amino acid and carbon metabolism in human adenocarcinoma cells (94). This creates an energy deficit, weakening the heart’s ability to maintain normal contractile and metabolic function, as in cardiac aging (3). Furthermore, infected human T-cells exhibit a metabolic shift toward glycolysis driven by enhanced mTOR signaling (95). This mirrors metabolic changes seen in aged hearts where reduced mitochondrial efficiency forces reliance on less efficient energy pathways, like glycolysis (96). Collectively, these alterations establish a self-perpetuating cycle of energy failure and oxidative stress which may lead to premature cardiac aging. HHV-6 infection may result in accelerated structural and functional cardiac aging. Recent clinical evidence revealed that HHV-6 infection induces cardiac arrhythmias, ventricular dilatation, and infiltration of immune cells, such as lymphocytes and neutrophils, within the myocardium of affected patients (97, 98). Persistent HHV-6 infection has also been linked to reductions in left ventricular ejection fraction (LVEF) in patients (99), indicating a progressive decline in cardiac performance. Importantly, the pathological influence of HHV-6 extends beyond the heart. Alongside related herpesviruses such as Epstein-Barr virus (EBV), HHV-6 has been implicated in systemic inflammatory conditions, including multiple sclerosis (100), which in turn increase susceptibility to age-associated vascular disorders such as acute coronary syndromes and cerebrovascular disease (101). HHV6 also interferes with autophagy in human T-cells primarily through the activation of mTOR signaling (95), which naturally becomes overactive with age as autophagy declines (3). By sustaining high mTOR activity, HHV6 effectively locks the cell in a dysfunctional, aged-like state. Evidence of this autophagic blockade includes the accumulation of sequestosome 1 (p62), a marker of impaired protein degradation, in donor peripheral blood mononuclear cells (PBMCs) (102). As p62 and misfolded or ubiquitinated proteins build up, they contribute to proteotoxic stress, a key driver of cardiac aging and pathology seen in dilated and ischemic cardiomyopathies (103). Thus, HHV6-induced autophagy inhibition not only mirrors natural aging mechanisms but also amplifies cellular dysfunction within the heart. Taken together, HHV-6 infection promotes cardiac aging through converging mechanisms that span myocardial inflammation, systemic immune dysregulation, mitochondrial dysfunction, and impaired proteostasis. By chronically activating mTOR signaling and inhibiting autophagy, HHV-6 enforces an aged cellular phenotype characterized by proteotoxic stress and declining contractile function. These viral-driven processes resemble and intensify age-related cardiac decline, ultimately increasing vulnerability to heart failure and vascular disease.
Coxsackievirus B. Coxsackievirus B (CVB) is a significant cardiotropic enterovirus strongly associated with human heart diseases, including myocarditis, pericarditis, and DCM (15). The mechanisms of CVB pathology, specifically mitochondrial disruption, subversion of autophagy, and induction of autoimmunity, create a state of chronic damage and dysfunction that accelerates cardiac aging. CVB directly targets the cellular systems that sustain energy production and mitochondrial quality control, accelerating the metabolic and structural decline typical in the aging heart. CVB3 infection induces upregulation of transferrin receptor protein 1, which disrupts cristae structure and increases membrane density in HeLa cells (104). It has been shown that altered mitochondrial morphology (including abnormal, irregularly arranged cristae and increased vacuole number) precedes cardiac inflammation in CVB3 infection of mice and primary mouse cardiomyocytes (105). Furthermore, CVB3 infection decreased ATP production, increased ROS production, and decreased membrane potential in infected rats and neonatal rat cardiomyocytes (105). These alterations are associated with decreased membrane potential, mitochondrial fragmentation, and an induction of autophagosome formation and mitophagy in infected HeLa cells (106). However, the decrease in membrane potential could not be replicated in H9C2 rat cardiomyocytes except at excessively high multiplicities of infection (MOI 50 or greater)(106). Conversely, one study showed an increase in respiration and ATP production which was dependent on ISG15, a target of interferon signaling, which suppresses viral replication and shifts cardiac metabolism toward oxidative phosphorylation in mice (107). The two studies use different strains of mice, so it is possible that genetic determinants of interferon signaling and susceptibility to subversion are determinants of susceptibility to viral myocarditis and DCM. The sustained immune response following CVB infection drives long-lasting cardiac pathology that persists even after the virus itself is cleared, leading to chronic age-like decline of the heart. CVB infection triggers an autoimmune response in patients in which antibodies generated against viral proteins cross-react with host cardiac proteins such as myosin, a key component of the contractile machinery (15). This phenomenon fuels autoimmune myocarditis, setting off a cycle of inflammation and tissue injury that progresses from acute myocarditis to long-term DCM and eventual heart failure (15, 108). Autophagy declines naturally with age, and CVB3 exploits this vulnerability to accelerate cardiac senescence. CVB3 disrupts protein clearance in HeLa cells by cleaving key autophagy-related proteins such as sequestosome 1 (p62) which normally aggregates ubiquitinated proteins and delivers them to autophagosomes for degradation (109–111). This disruption leads to the accumulation of ubiquitinated and misfolded proteins, forming toxic aggregates implicated in age-related cardiac pathologies such as dilated and hypertrophic cardiomyopathies and myocardial ischemia (103, 112). Autophagy protects the heart under stress, and its impairment by CVB3 resembles the dysfunction of an aged myocardium. Beyond general autophagy, CVB3 also hijacks mitophagy with devastating effects on cardiac metabolism. The virus induces mitophagy through the Parkin pathway in mice (113), but redirects autophagosomes to serve as replication niches and vehicles for viral egress rather than for degradation in mouse cardiomyocytes (114–116). This subversion may release damaged mitochondria and mitochondrial DNA (mtDNA) into the extracellular space (117), where mtDNA acts as a potent danger signal that triggers chronic inflammation through macrophage activation and cytokine release (TNF-α and IL-6). Accumulated mitochondria further generate ROS and apoptotic factors like cytochrome C (3, 103), intensifying oxidative stress and inflammation. Collectively, these processes create a self-perpetuating cycle of cellular damage, metabolic collapse, and inflammaging, converting healthy cardiomyocytes into senescent-like cells and paving the way for fibrosis, chronic inflammation, and heart failure. In Fig. 2 we have summarized the effects of viral infections on cardiovascular aging.
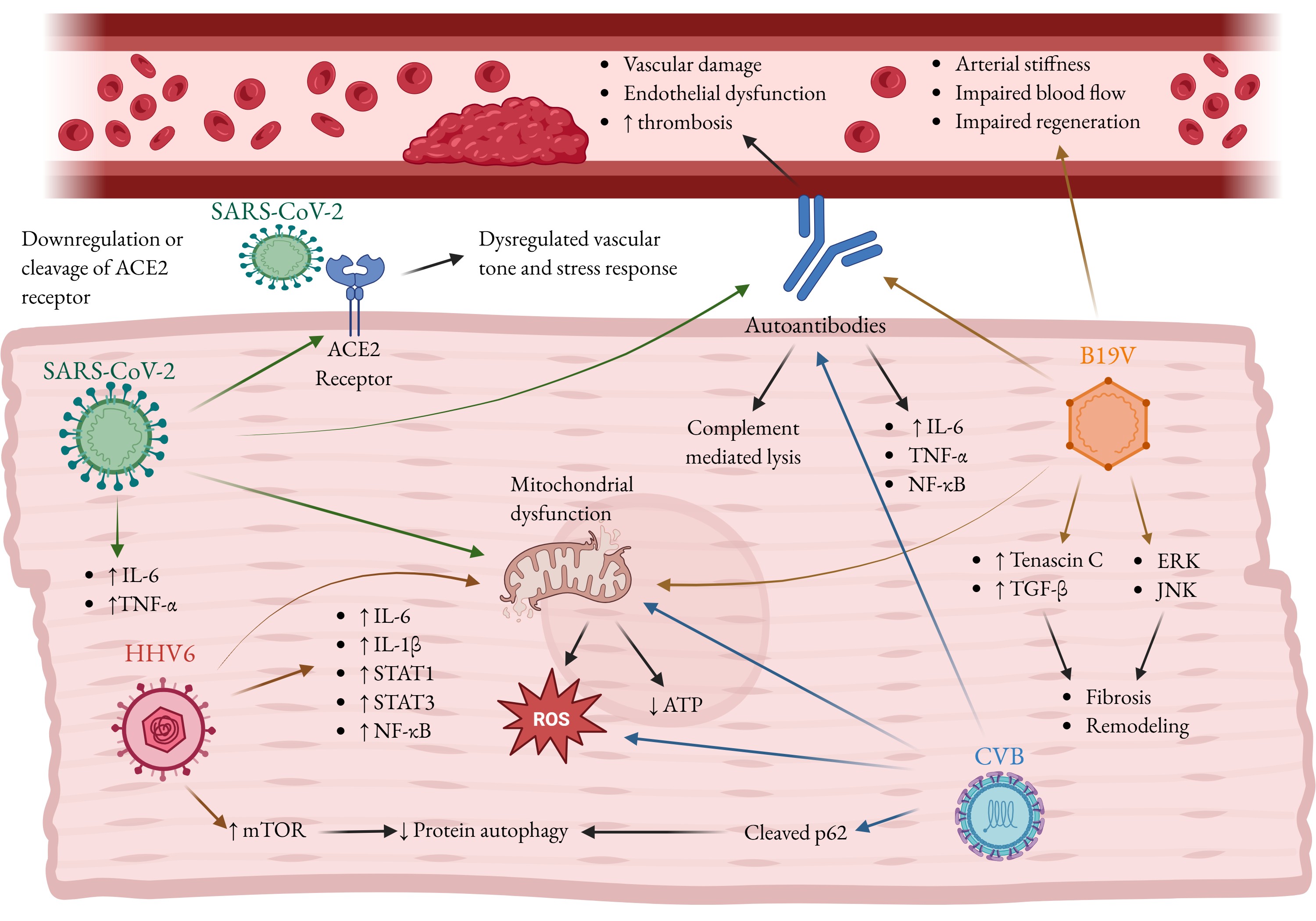
Fig. 2: Viral infections promote cardiac aging and disease. Shown above are SARS-CoV-2, Human herpusvirus-6 (HHV6), Human parvovirus B19 (B19V), Coxsackievirus (CVB) and the mechanisms by which they can contribute to an age-like diseased phenotype in the infected heart. Viral infection cause inflammation and, in the case of B19V, JNK and ERK signaling, which contribute to cardiac fibrosis and pathological tissue remodeling. Many viruses can cause mitochondrial dysfunction, an increase in ROS production and oxidative stress while decreasing ATP reserves, contributing to senescence. SARS-CoV-2 can cause cardiac dysfunction via binding to its viral entry receptor, angiotensin-converting enzyme 2 (ACE2). HHV6 increases mTOR signaling, while CVB cleaves sequestosome 1 (p62). Both inhibit proper autophagy, which contributes to aging and fibrosis. SARS-CoV-2 and B19V cause autoantibody formation, leading to endothelial dysfunction and thrombosis. Autoantibodies also cause inflammation and cell lysis via complement activation. B19V additionally infects endothelial cells, impairing blood flow and arterial regeneration, while promoting arterial stiffness. Fig. created by BioRender.
Parasitic infections
Parasitic infections, including Toxoplasma gondii, Trypanosoma cruzi, and Trichinella spiralis, elicit robust inflammatory immune responses and characteristically progress from an acute phase to chronic persistence. Many of these parasites exhibit cardiac tropism, while in other cases sustained systemic and local inflammation leads to collateral myocardial injury. Regardless of the initiating mechanism, chronic parasitic infection imposes prolonged inflammatory and metabolic stress on cardiomyocytes, thereby exacerbating age-associated cellular exhaustion and accelerating pathological cardiac remodeling. In the following sections, we highlight the molecular and cellular mechanisms through which these chronic parasitic infections promote cardiac aging and functional decline.
Toxoplasma gondii. Toxoplasma gondii is an obligate intracellular parasite infecting approximately one-third of the global population (118). The parasite utilizes three forms, tachyzoite (rapidly dividing), bradyzoite (tissue cyst), and oocyst (transmission/persistence), and is primarily transmitted through the ingestion of oocysts from contaminated food, water, or soil, or via tissue cysts in undercooked meat (119). Accumulating evidence links chronic T. gondii infection to accelerated cardiovascular disease and aging. The primary link between chronic T. gondii infection and cardiovascular disease appears to be sustained immune activation that drives excessive production of ROS, a key mediator of vascular aging. Persistent T. gondii infection promotes a prolonged Th1-dominant inflammatory response marked by elevated levels of IFN-γ, TNF-α, and IL-1β which stimulate ROS generation through mitochondrial dysfunction and activation of NADPH oxidases in immune and endothelial cells from humans and mice (120–122). Sustained oxidative burden overwhelms antioxidant defenses leading to redox imbalance, endothelial dysfunction, and cumulative vascular injury (123). This chronic ROS-rich inflammatory environment closely parallels, and amplifies, inflammaging, thereby accelerating vascular aging and increasing susceptibility to cardiovascular disease. Consistent with this mechanism, latent T. gondii infection is associated with elevated circulating biomarkers of endothelial injury vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) in chronic T. gondii-infected individuals reflecting persistent endothelial activation (124). Also, elevated serum levels of endothelin-1, a marker of endothelial dysfunction, have been reported in patients with acute toxoplasmosis (125). Moreover, acute T. gondii infection in mice demonstrates direct vascular involvement, inducing endothelial activation, upregulation of adhesion molecules, and structural alterations in cerebral vasculature (126). These changes are accompanied by vascular occlusion and impaired cerebral blood flow indicating functional vascular decline (126). At the cellular level, acute T. gondii infection of human umbilical vein endothelial cells (HUVECs) induces DNA damage, S-phase cell-cycle arrest, and cytokinesis failure (127). Although ROS are a well-recognized driver of DNA damage, intrinsic ROS production in infected HUVECs does not appear to account for the early DNA damage observed during infection (127). Importantly, these in vitro studies do not account for extrinsic sources of oxidative stress, such as ROS generated by infected macrophages. Therefore, future investigations should assess endothelial injury in more physiologically relevant settings that incorporate immune-endothelial interactions to better define the contribution of T. gondii infection to vascular aging. Epidemiological and clinical evidence consistently demonstrate that T. gondii infection is associated with increased cardiovascular risk similar to advanced age. Infected individuals frequently exhibit an adverse biomarker profile including elevated blood pressure, plasma triglycerides, and C-reactive protein (CRP), coupled with reduced HDL cholesterol, markers that typically worsen with age (128). Population-based studies suggest chronic subclinical cardiac involvement. Notably, case-control studies identified a strong association between T. gondii seropositivity and cardiovascular diseases, particularly in older adults (129, 130). In addition, another study found that T. gondii infection is associated with higher risk of cardiovascular mortality in men (131). Collectively, these findings indicate that T. gondii infection may act as a biological catalyst for cardiovascular aging by sustaining chronic inflammation and oxidative stress, which compromise vascular integrity and accelerate atherosclerosis.
Trypanosoma cruzi. Trypanosoma cruzi, an intracellular protozoan parasite, is the causative agent of Chagas disease. Chagas diseases is known to be endemic throughout Latin America, however a recent CDC report indicates endemic circulation of the disease in the USA as well (134). Chagas disease is transmitted predominantly through triatomine insects (kissing bugs) in rural regions where housing conditions favor vector colonization. However, non-vectorial routes such as congenital transmission, blood transfusion, organ transplantation, and ingestion of contaminated food or beverages also contribute to disease spread (132, 135). Following a 1-2 week incubation period, the acute phase typically lasts 4-8 weeks and is often marked by mild, nonspecific symptoms such as fever and malaise (136). If untreated, approximately 20-30% of infected individuals develop chronic Chagas cardiomyopathy (CCC), manifesting as progressive cardiac dysfunction (137, 138). Older individuals are particularly vulnerable, as T. cruzi infection in this group is associated with the gradual emergence of new electrocardiographic abnormalities and an increased risk of cardiac-related mortality (139, 140). CCC represents the most severe manifestation of the chronic phase of T. cruzi infection, characterized by persistent parasitic presence and a sustained dysregulated immune response, despite the typically low number of detectable parasites within cardiac tissue. Clinically, CCC presents with a spectrum of debilitating cardiac manifestations including conduction abnormalities like right bundle branch block, arrhythmias, ventricular aneurysms, heart failure, and an elevated risk of sudden cardiac death (137, 138). Mechanistically, T. cruzi infection initiates a destructive cycle of immune activation, inflammation, oxidative stress, and mitochondrial dysfunction in the hearts of patients (141, 142) that closely resembles cardiac aging (2). Upon T. cruzi infection, parasite-derived molecules, including glycosylphosphatidylinositol (GPI) anchors and nucleic acids, are recognized by host Toll-like receptors (TLRs), particularly TLR2 and TLR9, initiating MyD88-dependent signaling cascades that activate NF-κB (143). This signaling axis drives the production of key proinflammatory mediators via IL-17 signaling, including IL-12, TNF-α, and IFN-γ which are essential for the development of effective innate and adaptive immune responses and play a critical role in host resistance to T. cruzi infection (143, 144). Also, it has been shown robust production of IL-17 promotes Th17 responses which aid resistance against infection and reduce cardiac inflammation in mice (144). IFN-γ enhances macrophage activation and the generation of reactive oxygen and nitrogen species (ROS and RNS) through induction of inducible nitric oxide synthase and NADPH oxidase, which are essential for parasite control, but also contribute to collateral oxidative damage in host tissues, including the myocardium (145, 146). Persistent ROS/RNS production in T. cruzi infected patients has been linked to oxidative injury of cardiac lipids, proteins, and DNA, and subsequent functional impairment of the heart. Persistent oxidative and inflammatory stress ultimately drives myocardial senescence, fibrosis, and remodeling leading to contractile dysfunction in rats and H9C2 rat cardiomyocytes (147). Chronic T. cruzi infection maintains elevated pro-inflammatory cytokines (IFN-γ, TNF-α, IL-6, and IL-1β) in CCC patients, correlating with myocardial damage severity, whereas higher IL-10 levels are linked to better outcomes (148). A key mechanism underlying this immune-mediated pathology is resemblance of T. cruzi antigens to its host cells. This can result in a persistent Th1-dominant inflammatory environment promoting autoreactivity against cardiac tissue. Recent studies demonstrate that specific alleles of HLA-DRB1, a type of immune molecule that helps present protein fragments to T cells, found in patients can present peptides with sequence homology between T. cruzi antigens and host cardiac myosin (149). This leads to the activation of autoreactive CD4⁺ T cells in CCC patients and enhanced IFN-γ production and cytotoxic gene expression, which are strongly associated with ventricular dysfunction and myocardial fibrosis (149). During T. cruzi infection in humans, the immunoregulatory protein galectin-1 (Gal-1) is upregulated and affects cardiac pathology and immune responses (151). It has two distinct context-dependent functions and is upregulated in infected tissues, particularly the heart (152). On the one hand, Gal-1 helps to limit acute tissue damage by protecting mouse cardiomyocytes from parasite infection and parasite-induced cell death (152). However, by stimulating regulatory T cells (CD4⁺CD25⁺Foxp3⁺) and raising anti-inflammatory mediators like IL-10 and TGF-β1, it inhibits Th1 and CD8⁺ T cell responses to the parasite in mice (151, 153). This immune suppression lessens the host's capacity to eradicate the infection and promotes parasite persistence. Gal-1 has dose-dependent effects in mouse macrophages; at low concentrations, it reduces microbicidal activity and promotes parasite survival; at higher concentrations, it triggers macrophage apoptosis and inhibition of parasite replication (151). Overall, Gal-1 contributes to the complex immunopathology of Chagas disease as well as parasite persistence by balancing the protection of cardiac tissue with the suppression of effective anti-parasite immunity. Elevated levels of IFN-γ and TNF-α are central to the pathogenesis of CCC, driving sustained activation of NF-κB signaling and upregulation of inducible nitric oxide synthase (iNOS) in patient hearts (141, 142). This inflammatory cascade leads to excessive production of nitric oxide (NO), ROS, and the potent oxidant peroxynitrite within patient cardiomyocytes, resulting in widespread oxidative and nitrosative damage (141, 142). The ensuing oxidative stress disrupts mitochondrial membrane potential, impairs ATP synthesis, and causes depletion of mtDNA, as demonstrated in both the human CCC myocardium and cytokine-treated cardiomyocytes (141, 142, 154). Electron leakage and an overabundance of mitochondrial reactive oxygen species (mtROS) may be caused by an electron transport chain disruption seen in mouse cardiomyocytes (155). The NFE2L2-antioxidant response element pathway is inhibited by the persistent production of mtROS in mice, impairing the antioxidant defenses of the cell and encouraging oxidative adduct accumulation, myocardial fibrosis, and left ventricular dysfunction (156). This persistent mitochondrial dysfunction sustains oxidative stress and energy deficit, resulting in a metabolic profile marked by reduced mitochondrial metabolism and increased immune activation closely resembling age-related heart failure (141, 142, 154, 157). T. cruzi infection provides a powerful model for understanding how chronic infections can accelerate cardiac aging, emphasizing the need for therapeutic strategies that simultaneously target persistent infection, metabolic dysfunction, and chronic inflammation.
Trichinella spiralis. Trichinella spiralis is a globally distributed zoonotic nematode responsible for trichinellosis with an estimated global incidence of around 10,000 human cases per year (158). Transmission occurs primarily through the consumption of raw or undercooked meat containing infectious larvae (159). T. spiralis infection offers a compelling case of how an infectious agent can initiate and exacerbate severe cardiac pathology. In a significant proportion of infected individuals, an estimated 10 to 60%, cardiovascular complications emerge, including eosinophilic myocarditis, pericarditis, and thrombotic events (160, 161). Among these, eosinophilic myocarditis is particularly consequential, often accompanied by elevated cardiac troponin levels and characteristic electrocardiographic abnormalities (161). While T. spiralis infection initially triggers acute myocarditis, in the long term it induces a chronic pathological state via immune suppression which resembles inflammaging. Cardiac damage is driven in large part by intense eosinophil infiltration and the release of granule proteins, which perpetuate myocardial inflammation and tissue remodeling in mice (162). T. spiralis also employs sophisticated immune evasion strategies, inducing a shift in mouse macrophage populations from pro-inflammatory M1 to anti-inflammatory M2 phenotypes (163, 164), and suppressing pro-inflammatory T cells (165). Although this immunosuppressive shift mitigates overwhelming acute inflammation, it may facilitate parasite persistence and sustain chronic, low-grade inflammation. A central mediator connecting T. spiralis infection to cardiac aging is Galectin-3 (Gal-3), a key regulator of inflammation and fibrosis. Gal-3 is a recognized biomarker and effector molecule in age-related cardiac remodeling and heart failure (166, 167). During acute T. spiralis infection in mice, Gal-3 expression rises markedly, although inhibition of galectin receptor interactions exacerbates myocardial inflammation, dysfunction, and fibrosis (162). These findings suggest that Gal-3 may function as a compensatory mechanism, acting to limit excessive inflammatory damage and fibrotic remodeling in the infected myocardium. Furthermore, T. spiralis infection perturbs host lipid metabolism in mice, particularly glycerophospholipid pathways (168), while Gal-3 itself promotes hepatic lipid accumulation in mice (169). The convergence of these effects, elevated Gal-3 signaling and disrupted lipid metabolism, synergistically drives fibrotic remodeling and cardiac hypertrophy in hamsters and H9C2 rat cardiomyocytes (162, 170). Together, these processes create a metabolic and structural environment that closely mirrors the progressive decline of the aging heart, underscoring how infection-induced immune and metabolic dysregulation may emulate and accelerate cardiac aging. Fig. 3 summarizes the effects of parasitic infections on cardiovascular aging.
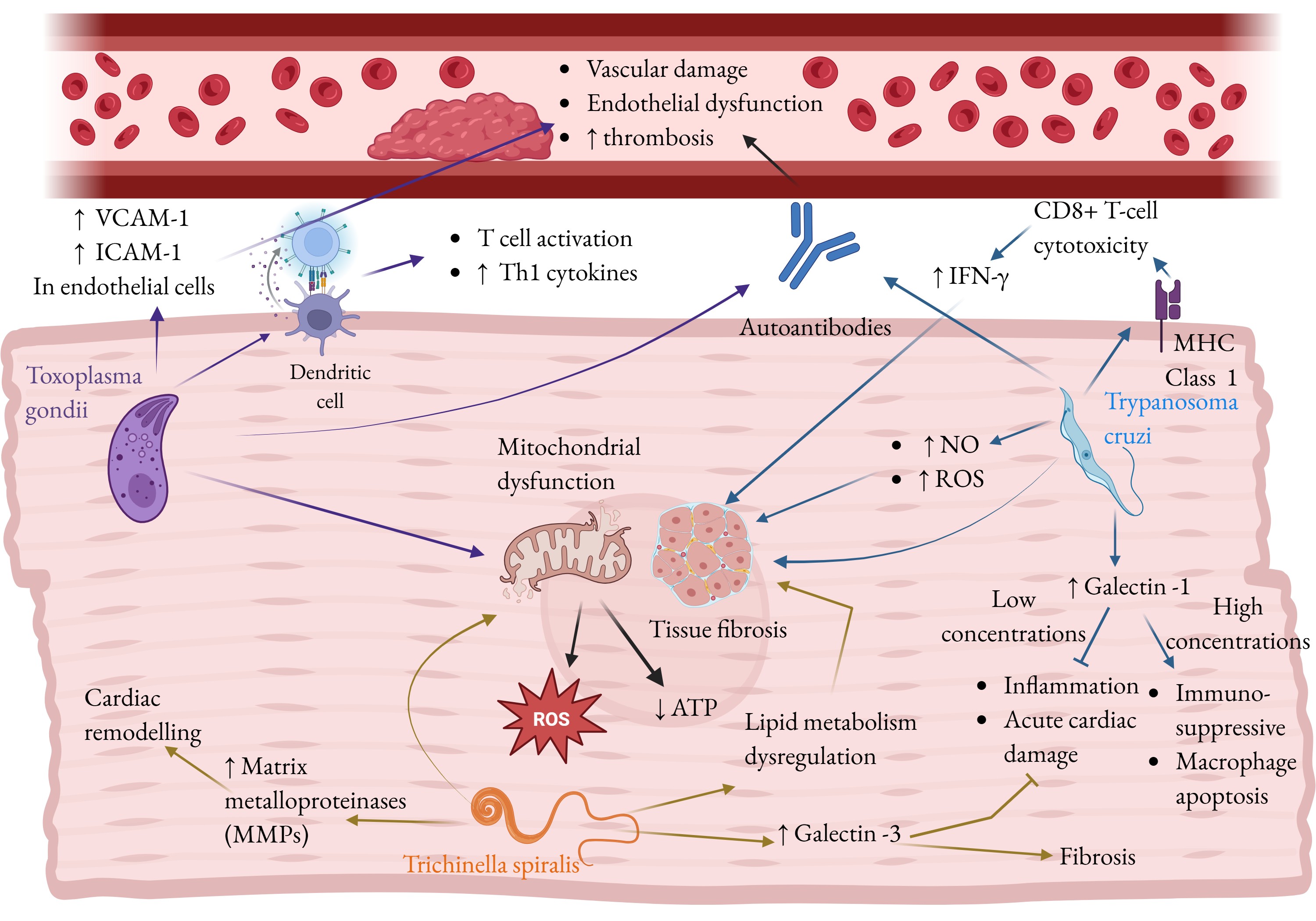
Fig. 3: Parasitic infections promote cardiac aging and disease. The parasites Toxoplasma gondii, Trypanosoma cruzi, and Trichinella spiralis are shown alongside the pathways by which they cause an aged-like diseased phenotype in the heart. Parasitic infections promote inflammation via various pathways, including dendritic cell activation and autoantibody production in T. gondii, MHC class 1 upregulation and autoantibody formation in T. cruzi, and cytokine production in T. spiralis. This inflammation promotes fibrosis in the heart. Autoantibody production also leads to endothelial damage and dysfunction, and thrombosis. T. gondii and T. spiralis cause mitochondrial dysfunction which increases reactive oxygen species (ROS) production and decreases ATP reserves, contributing to senescence. T. gondii decreases nitric oxide production, leading to myocardial stiffening, and VCAM-1, a marker of endothelial activation. T. cruzi and T. spiralis increase galectin signaling, which decrease acute inflammation and cardiac damage, but suppress the immune response to allow parasite persistence, and promote fibrosis. Additionally, T. spiralis upregulates matrix metalloproteinases (MMPs) which mediate pathological cardiac remodeling. Fig. created by BioRender.
Bacterial Infections
Bacterial infections can promote cardiac aging by inducing the release of cytokines causing damage to cardiac cells. Bacterial infections such as Staphylococcus aureus, Streptococcus pneumoniae, and Mycoplasma pneumoniae contribute to fibrosis of heart tissue, weakening heart muscle function as in cardiac aging.
Staphylococcus aureus.Staphylococcus aureus is a critical Gram-positive pathogen responsible for severe and often life-threatening cardiovascular infections including myocarditis and infective endocarditis (IE). These conditions rank among the most fatal cardiac infections with IE exhibiting a particularly high mortality rate (171, 172). The pathology and severity of S. aureus infections are further intensified by the natural process of cardiac aging, which reduces tissue regeneration and immune competence (42, 173). S. aureus employs a range of virulence factors that are especially destructive to the aging cardiovascular system. For instance, S. aureus secreted enterotoxins are known to activate host inflammatory pathways inducing multiple forms of programmed cell death including apoptosis, pyroptosis, and necroptosis (174, 175). The pore-forming α-toxin targets epithelial and endothelial cells, leukocytes, and platelets (176, 177) promoting chronic endothelial injury in patients that facilitates thrombogenesis and the formation of a mass of proliferating bacteria shielded by serum molecules and platelets called a vegetation (178). Moreover, S. aureus utilizes antibiotic-neutralizing enzymes that enhance antimicrobial resistance resulting in dangerous antibiotic resistant strains (179). S. aureus infection may lead to infective endocarditis, a disease increasingly associated with advanced age. S. aureus is the most frequent pathogen identified in infective endocarditis, as demonstrated in a cohort study of 2, 781 patients where it accounted for 31.2% of cases and was commonly linked to mitral and aortic valve damage, heart failure, and complications such as embolization, pulmonary edema, and stroke (180). The rising incidence of infective endocarditis among elderly populations is partly attributed to degenerative valvular diseases, such as calcific stenosis, that provide a damaged surface conducive to bacterial colonization (180). Moreover, S. aureus infection is associated with a higher risk of in-hospital mortality, particularly in patients with advanced age, prosthetic valve involvement, or paravalvular complications (180). A study of 221 patients with confirmed infective endocarditis following S. aureus bacteremia found vegetations in 65.9% of cases. These vegetations, attached to the endocardium, underscore the rapid progression of the infection (181). Beyond infective endocarditis, S. aureus is also the most common bacterial cause of myocarditis in humans characterized by the formation of small abscesses, typically in the left ventricle (182). S. aureus myocarditis results in cardiac dysfunction, arrhythmias, myocardial rupture, and purulent pericarditis in infected patients (182). In the aged myocardium, where regenerative capacity is diminished (173), these structural complications are both more frequent and more severe. Methicillin-resistant Staphylococcus aureus (MRSA) refers to highly virulent and drug-resistant strains associated with more severe complications. Clinically, MRSA endocarditis leads to significantly worse outcomes than methicillin-susceptible S. aureus infections including higher rates of renal complications, persistent bacteremia, and mortality (183). Alarmingly, MRSA-related deaths in the United States have surpassed those attributed to AIDS, Parkinson’s disease, and homicide (184). Mechanistically, MRSA’s resistance to β-lactam antibiotics is mediated by the mecA gene which encodes the altered penicillin-binding protein PBP2a (184, 185). Treatment is further challenged by the need for alternative antibiotics such as vancomycin or daptomycin (186), and by the emergence of vancomycin-intermediate and vancomycin-resistant S. aureus strains (187). Moreover, MRSA biofilms on mechanical cardiac valves and implanted medical devices perpetuate chronic infection (188). In older adults who are more likely to have implanted devices, this biofilm persistence contributes to recurrent bacteremia and often necessitates aggressive management, including surgical intervention (188).
Streptococcus pneumoniae. Streptococcus pneumoniae, a common opportunistic pathogen, has emerged as a significant contributor to cardiovascular disease. The mechanisms of S. pneumoniae-induced cardiotoxicity directly intersect with and accelerate cardiac aging by amplifying tissue injury and exploiting age-related vulnerabilities. A key driver of this process is pneumolysin, a pore-forming toxin that induces human and mouse cardiomyocyte death via necroptosis (189–191). Necroptosis is a highly proinflammatory form of programmed cell death mediated by RIPK1 and RIPK3 activation, followed by MLKL-dependent membrane disruption, ion imbalance, and cellular energy loss (192). In the aged heart, already characterized by reduced cardiomyocyte reserves, heightened oxidative stress, and chronic low-grade inflammation (41, 42, 173, 193), this necroptosis-mediated cell loss can impose a greater burden. In later stages of infection, disrupted cellular membranes release alarmins which amplify inflammation (194). The release of inflammatory signals such as alarmins causes inflammatory damage to the heart leading to the development of heart failure or higher mortality in existing heart failure (16, 17, 195). Moreover, pneumolysin-induced necroptosis drives long-term cardiac remodeling, marked by de novo collagen deposition (fibrosis) and reduced fractional shortening in mice and in human and mouse cardiomyocytes (191). Fibrosis and stiffness are hallmarks of the aging myocardium, positioning S. pneumoniae infection as an acute insult that hastens the heart’s structural and functional transition toward a senescent phenotype. S. pneumoniae infection is closely linked to acute cardiovascular complications, which occur more frequently and with greater severity in older individuals. Clinical evidence indicates that S. pneumoniae infection significantly increases the risk of coronary heart disease, heart failure, stroke, and atrial fibrillation (196), with hospitalizations for pneumococcal disease often resulting from new or worsened heart failure and cardiac arrhythmias (197). The pathogen’s success in establishing infection and driving cardiac injury is partly due to immune suppression. During the early stages of infection, pneumolysin suppresses the production of key pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6, and IL-12 by inducing suppressor of cytokine signaling 1 (SOCS1) in human dendritic cells and mouse macrophages, which inhibits downstream immune signaling and weakens host defense activation (190, 198). This early immune suppression is particularly harmful in the elderly, where immune senescence limits the capacity for efficient immune activation (199–201). As a result, S. pneumoniae gains an early advantage allowing unchecked bacterial proliferation before the onset of destructive late-stage inflammation that further damages cardiac tissue. The localized cardiac damage caused by S. pneumoniae underscores its ability to induce chronic, age-like structural changes in the heart. Studies in humans, mice, and rhesus macaques have identified distinct myocardial microlesions during S. pneumoniae infection (202), while in vitro experiments show that even at a low multiplicity of infection, approximately 60% of HL-1 mouse cardiomyocytes undergo cell death within just six hours of exposure in vitro (197). Although pneumococcal endocarditis and aortitis are relatively uncommon, they progress rapidly when they occur leading to massive thrombus formation and severe valve destruction, most frequently affecting the aortic valve and often necessitating early surgical intervention (203). In non-human primate models, myocardial injury has been confirmed by elevated levels of troponin T and heart-type fatty acid-binding protein accompanied by persistent cardiac scarring even after successful antibiotic therapy (204). This enduring structural damage demonstrates that S. pneumoniae-induced cardiac injury extends well beyond the acute infectious phase, leaving behind fibrotic and functional alterations, thereby highlighting the need for therapeutic strategies beyond standard antibiotic treatment.
Mycoplasma pneumoniae. Mycoplasma pneumoniae is a small, self-replicating pathogen best known for causing community-acquired pneumonia. Beyond its respiratory tropism, it has the capacity to induce severe multisystemic disease including cardiovascular complications such as pericarditis, myocarditis, congestive heart failure, and, in rare cases, infective endocarditis (205). The mechanisms underlying M. pneumoniae’s extrapulmonary effects, spanning cardiovascular, neurological, and dermatological systems (206, 207), closely align with the biological processes that drive cardiac aging, particularly chronic inflammation. M. pneumoniae elicits a potent pro-inflammatory response marked by elevated levels of cytokines such as TNF-α, IFN-γ, IL-1β, IL-4, IL-6, IL-8, and IL-18 (208). In older individuals whose systems already experience inflammaging, this exaggerated immune activation overwhelms homeostatic control, exacerbates endothelial injury, and promotes cardiac remodeling. The resulting fibrosis and stiffness mirror the structural and functional decline characteristic of the aged heart. M. pneumoniae employs multiple immune-modulation and virulence strategies to enact inflammatory damage to the myocardium, leading to long-term functional decline. Through molecular mimicry, the pathogen produces proteins that closely resemble host molecules such as keratin, fibrinogen, vimentin, and various glycoproteins provoking autoimmunity in patients (209). This autoimmune activation is particularly detrimental to the heart, as chronic, misdirected immune attacks against cardiac tissues, such as the valves and pericardium, lead to age-like damage and fibrosis in Mycoplasma patients. Additionally, M. pneumoniae secretes the Community Acquired Respiratory Distress Syndrome (CARDS) toxin, which enters mammalian epithelial cells and modifies host proteins via ADP-ribosylation. This induces inflammation via the NLRP3-associated inflammasome, releasing IL-1β and IL-18 (209). This toxin-driven cellular injury compounds the existing burden of oxidative and replicative stress characteristic of aged tissues, further promoting senescence. Clinically, these pathogenic mechanisms manifest as severe cardiovascular involvement. M. pneumoniae has been documented as a rare cause of infective endocarditis in an immunocompetent adult, presenting with fever, elevated troponin-I levels indicative of myocardial injury, and vegetation on the pulmonary valve (205). It’s ability to sustain chronic inflammation, trigger oxidative injury, and provoke autoimmunity suggests that, beyond causing acute respiratory illness, M. pneumoniae exerts lasting pathological effects that may drive cardiovascular aging. In Fig. 4, we provide an overview of how bacterial infections contribute to cardiovascular aging.
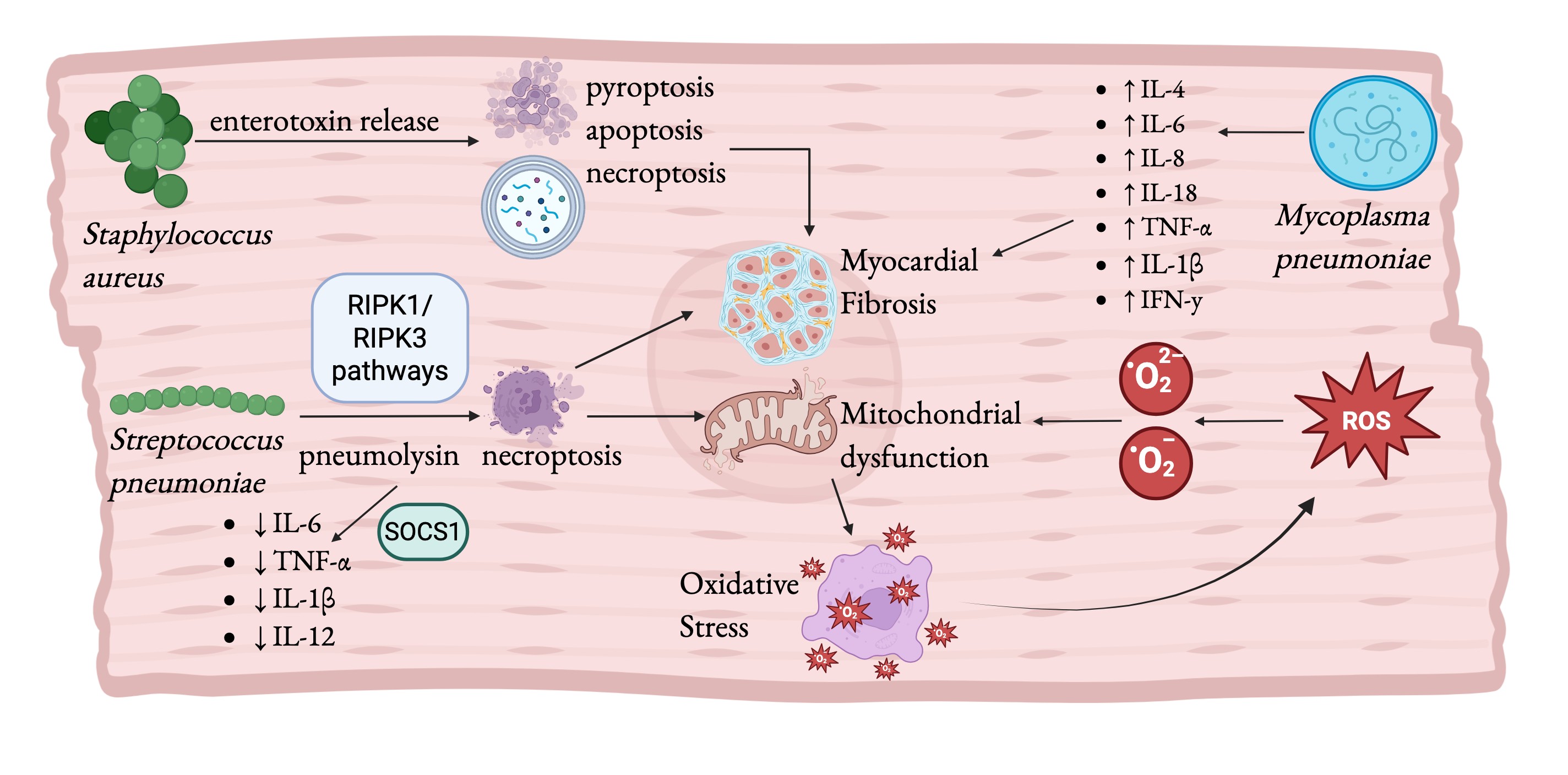
Fig. 4: Bacterial infections promote cardiac aging and disease. The pathogenic bacteria Staphylococcus aureus, Streptococcus pneumoniae, and Mycoplasma pneumoniae are shown alongside mechanisms by which they cause and aged-like diseased phenotype. Bacterial infections cause widespread inflammation, including the release of enterotoxin and pneumolysin. These toxins trigger programed cell death which increases inflammation and leads to fibrosis. M. pneumoniae increases reactive oxygen species (ROS) production, contributing to senescence. ROS and necroptosis caused by M. pneumoniae and S. pneumoniae respectively cause mitochondrial dysfunction, a hallmark of the aging heart that further contributes to oxidative stress. Fig. created by BioRender.
Concluding remarks
Cardiovascular aging is a multifaceted process driven by a complex interplay of cellular, molecular, and systemic factors that culminate in structural remodeling and functional decline of the heart. Among the key contributors, chronic inflammation and oxidative stress emerge as central mechanisms that accelerate tissue injury, impair repair pathways, and promote fibrosis and metabolic imbalance. Increasing evidence indicates that chronic infections, including viral, parasitic, and bacterial pathogens, can profoundly influence cardiovascular aging. By establishing persistent inflammatory and oxidative environments, these infections exacerbate mitochondrial dysfunction, endothelial damage, and maladaptive immune responses, thereby accelerating age-associated cardiac deterioration. Such processes contribute to a spectrum of cardiovascular pathologies, including atherosclerosis, DCM atrial fibrillation, and myocardial infarction. Conversely, the aging cardiovascular system itself becomes increasingly susceptible to chronic infection. Age-related declines in immune resilience, redox balance, and cellular repair mechanisms create a permissive environment for persistent pathogens to thrive and inflict further damage. This bidirectional relationship between chronic infection and cardiovascular aging represents a critical yet underexplored dimension of heart disease pathogenesis. Understanding the molecular networks that link infection, inflammation, and aging offers an opportunity to identify novel biomarkers and therapeutic targets that could preserve cardiac resilience, slowing the trajectory of age-related cardiovascular decline. Future studies integrating systems-level, multi-omics, and spatial approaches are essential to unravel the regional and cell-type-specific vulnerabilities of the aging heart. Such efforts will not only deepen our mechanistic understanding of cardiovascular aging, but also inform strategies to promote healthy cardiac longevity in the context of chronic infectious and inflammatory stressors.
Disclosure Statement
The authors declare that they have no competing financial interests or personal relationships that could have influenced the work reported in this manuscript.
References
- Ahmad FB, Cisewski JA, Anderson RN. Mortality in the United States — Provisional Data, 2023. MMWR Morb Mortal Wkly Rep. 2024 Aug 8;73(31):677–81 doi:10.15585/mmwr.mm7331a1
- Abdellatif M, Rainer PP, Sedej S, Kroemer G. Hallmarks of cardiovascular ageing. Nat Rev Cardiol. 2023 Nov;20(11):754–77. doi:10.1038/s41569-023-00881-3
- Yan M, Sun S, Xu K, Huang X, Dou L, Pang J, et al. Cardiac Aging: From Basic Research to Therapeutics. Oxid Med Cell Longev. 2021;2021:9570325. doi:10.1155/2021/9570325 PubMed PMID: 33777324; PubMed Central PMCID: PMC7969106.
- Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation. 2003 Jan 21;107(2):346–54. doi:10.1161/01.cir.0000048893.62841.f7 PubMed PMID: 12538439.
- Headley C, Turner J, Rajaram MV. Aging heart and infection. Aging. 2019 Jul 25;11(14):4781–2. doi:10.18632/aging.102128 PubMed PMID: 31346150; PubMed Central PMCID: PMC6682520.
- Johnson A, Rought T, Aronov J, Pokharel P, Chiu A, Nasuhidehnavi A. The impacts of chronic infections on shaping cellular senescence. Immun Ageing. 2025 Oct 10;22(1):37. doi:10.1186/s12979-025-00533-9
- Carrillo-Salinas FJ, Ngwenyama N, Anastasiou M, Kaur K, Alcaide P. Heart Inflammation. Am J Pathol. 2019 Aug;189(8):1482–94. doi:10.1016/j.ajpath.2019.04.009
- Badrinath A, Bhatta S, Kloc A. Persistent viral infections and their role in heart disease. Front Microbiol. 2022;13:1030440. doi:10.3389/fmicb.2022.1030440 PubMed PMID: 36504781; PubMed Central PMCID: PMC9730422.
- Mukherjee A, Ghosh KK, Chakrabortty S, Gulyás B, Padmanabhan P, Ball WB. Mitochondrial Reactive Oxygen Species in Infection and Immunity. Biomolecules. 2024 Jun 8;14(6):670. doi:10.3390/biom14060670 PubMed PMID: 38927073; PubMed Central PMCID: PMC11202257.
- Le Sage V, Cinti A, Amorim R, Mouland AJ. Adapting the Stress Response: Viral Subversion of the mTOR Signaling Pathway. Viruses. 2016 May 24;8(6):152. doi:10.3390/v8060152 PubMed PMID: 27231932; PubMed Central PMCID: PMC4926172.
- Molnar T, Lehoczki A, Fekete M, Varnai R, Zavori L, Erdo-Bonyar S, et al. Mitochondrial dysfunction in long COVID: mechanisms, consequences, and potential therapeutic approaches. GeroScience. 2024 Apr 26;46(5):5267–86. doi:10.1007/s11357-024-01165-5
- Aguilera MO, Delgui LR, Romano PS, Colombo MI. Chronic Infections: A Possible Scenario for Autophagy and Senescence Cross-Talk. Cells. 2018 Oct 10;7(10):162. doi:10.3390/cells7100162
- Ewald S, Nasuhidehnavi A, Feng TY, Lesani M, McCall LI. The intersection of host in vivo metabolism and immune responses to infection with kinetoplastid and apicomplexan parasites. Lodoen MB, editor. Microbiol Mol Biol Rev. 2024 Mar 27;88(1):e00164-22. doi:10.1128/mmbr.00164-22
- Izzo C, Vitillo P, Di Pietro P, Visco V, Strianese A, Virtuoso N, et al. The Role of Oxidative Stress in Cardiovascular Aging and Cardiovascular Diseases. Life. 2021 Jan 15;11(1):60. doi:10.3390/life11010060 PubMed PMID: 33467601; PubMed Central PMCID: PMC7829951.
- Lasrado N, Reddy J. An overview of the immune mechanisms of viral myocarditis. Rev Med Virol. 2020 Nov;30(6):1–14. doi:10.1002/rmv.2131 PubMed PMID: 32720461.
- Frantz S, Falcao-Pires I, Balligand JL, Bauersachs J, Brutsaert D, Ciccarelli M, et al. The innate immune system in chronic cardiomyopathy: a European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur J Heart Fail. 2018 Mar;20(3):445–59 doi:10.1002/ejhf.1138 PubMed PMID: 29333691; PubMed Central PMCID: PMC5993315.
- Lafuse WP, Wozniak DJ, Rajaram MVS. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells. 2020 Dec 31;10(1):51. doi:10.3390/cells10010051 PubMed PMID: 33396359; PubMed Central PMCID: PMC7824389.
- Abdellatif M, Rainer PP, Sedej S, Kroemer G. Hallmarks of cardiovascular ageing. Nat Rev Cardiol. 2023 Nov;20(11):754–77. doi:10.1038/s41569-023-00881-3 PubMed PMID: 37193857.
- Shin HJ, Kim IS, Kim JK, Jo EK. Molecular mechanisms of NLRP3 inflammasome activation. Exp Mol Med. 2026 Mar 25;58(3):650–63. doi:10.1038/s12276-026-01656-9
- Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017 Jul 14;2(1):17023. doi:10.1038/sigtrans.2017.23
- Liu WB, Wang SS, Zhang X, Ke ZZ, Wen XY, Zhao J, et al. Enhanced Cardiomyocyte NLRP3 Inflammasome-Mediated Pyroptosis Promotes d-Galactose-Induced Cardiac Aging. J Am Heart Assoc. 2024 Jul 16;13(14):e032904. doi:10.1161/JAHA.123.032904 PubMed PMID: 38979831; PubMed Central PMCID: PMC11292767.
- Liao LZ, Chen ZC, Wang SS, Liu WB, Zhao CL, Zhuang XD. NLRP3 inflammasome activation contributes to the pathogenesis of cardiocytes aging. Aging. 2021 Aug 25;13(16):20534–51. doi:10.18632/aging.203435 PubMed PMID: 34432650; PubMed Central PMCID: PMC8436929.
- Tang X, Li PH, Chen HZ. Cardiomyocyte Senescence and Cellular Communications Within Myocardial Microenvironments. Front Endocrinol. 2020;11:280. doi:10.3389/fendo.2020.00280 PubMed PMID: 32508749; PubMed Central PMCID: PMC7253644.
- Liberale L, Badimon L, Montecucco F, Lüscher TF, Libby P, Camici GG. Inflammation, Aging, and Cardiovascular Disease. J Am Coll Cardiol. 2022 Mar;79(8):837–47. doi:10.1016/j.jacc.2021.12.017
- Morrisette-Thomas V, Cohen AA, Fülöp T, Riesco É, Legault V, Li Q, et al. Inflamm-aging does not simply reflect increases in pro-inflammatory markers. Mech Ageing Dev. 2014 Jul;139:49–57. doi:10.1016/j.mad.2014.06.005 PubMed PMID: 25011077; PubMed Central PMCID: PMC5881904.
- Ramos GC, Van Den Berg A, Nunes-Silva V, Weirather J, Peters L, Burkard M, et al. Myocardial aging as a T-cell–mediated phenomenon. Proc Natl Acad Sci. 2017 Mar 21;114(12). doi:10.1073/pnas.1621047114
- Ashour D, Rebs S, Arampatzi P, Saliba AE, Dudek J, Schulz R, et al. An interferon gamma response signature links myocardial aging and immunosenescence. Cardiovasc Res. 2023 Nov 15;119(14):2458–68. doi:10.1093/cvr/cvad068
- Li X, Bao Y, Zhang N, Lin C, Xie Y, Wei Y, et al. Senescent CD8+ T cells: a novel risk factor in atrial fibrillation. Cardiovasc Res. 2025 Apr 15;121(1):97–112. doi:10.1093/cvr/cvae222
- Frangogiannis NG. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med. 2019 Feb;65:70–99. doi:10.1016/j.mam.2018.07.001
- Bradshaw AD, DeLeon-Pennell KY. T-cell regulation of fibroblasts and cardiac fibrosis. Matrix Biol J Int Soc Matrix Biol. 2020 Sep;91–92:167–75. doi:10.1016/j.matbio.2020.04.001 PubMed PMID: 32438054; PubMed Central PMCID: PMC7434661.
- Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo-Salinas FJ, Aronovitz M, et al. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med. 2017 Nov 6;214(11):3311–29. doi:10.1084/jem.20161791 PubMed PMID: 28970239; PubMed Central PMCID: PMC5679176.
- Ramos GC, van den Berg A, Nunes-Silva V, Weirather J, Peters L, Burkard M, et al. Myocardial aging as a T-cell-mediated phenomenon. Proc Natl Acad Sci U S A. 2017 Mar 21;114(12):E2420–9. doi:10.1073/pnas.1621047114 PubMed PMID: 28255084; PubMed Central PMCID: PMC5373357.
- Xia G, Zhu S, Liu Y, Pan J, Wang X, Shen C, et al. Transcriptomic profiling and regulatory pathways of cardiac resident macrophages in aging. Cell Mol Life Sci CMLS. 2024 May 20;81(1):220. doi:10.1007/s00018-024-05235-x PubMed PMID: 38763956; PubMed Central PMCID: PMC11102896.
- Fang Z, Raza U, Song J, Lu J, Yao S, Liu X, et al. Systemic Aging Fuels Heart Failure: Molecular Mechanisms and Therapeutic Avenues. ESC Heart Fail. 2025 Apr 1;12(2):1059–80. doi:10.1002/ehf2.14947
- Frangogiannis NG, Dewald O, Xia Y, Ren G, Haudek S, Leucker T, et al. Critical Role of Monocyte Chemoattractant Protein-1/CC Chemokine Ligand 2 in the Pathogenesis of Ischemic Cardiomyopathy. Circulation. 2007 Feb 6;115(5):584–92. doi:10.1161/CIRCULATIONAHA.106.646091
- Van Amerongen MJ, Harmsen MC, Van Rooijen N, Petersen AH, Van Luyn MJA. Macrophage Depletion Impairs Wound Healing and Increases Left Ventricular Remodeling after Myocardial Injury in Mice. Am J Pathol. 2007 Mar;170(3):818–29. doi:10.2353/ajpath.2007.060547
- Zannas AS, Jia M, Hafner K, Baumert J, Wiechmann T, Pape JC, et al. Epigenetic upregulation of FKBP5 by aging and stress contributes to NF-κB-driven inflammation and cardiovascular risk. Proc Natl Acad Sci U S A. 2019 Jun 4;116(23):11370–9. doi:10.1073/pnas.1816847116 PubMed PMID: 31113877; PubMed Central PMCID: PMC6561294.
- Helenius M, Hänninen M, Lehtinen SK, Salminen A. Aging-induced up-regulation of nuclear binding activities of oxidative stress responsive NF-kB transcription factor in mouse cardiac muscle. J Mol Cell Cardiol. 1996 Mar;28(3):487–98. doi:10.1006/jmcc.1996.0045 PubMed PMID: 9011632.
- Zhu B, Zhang L, Liang C, Liu B, Pan X, Wang Y, et al. Stem Cell-Derived Exosomes Prevent Aging-Induced Cardiac Dysfunction through a Novel Exosome/lncRNA MALAT1/NF-κB/TNF-α Signaling Pathway. Oxid Med Cell Longev. 2019;2019:9739258. doi:10.1155/2019/9739258 PubMed PMID: 31089420; PubMed Central PMCID: PMC6476062.
- Gupta S, Young D, Maitra RK, Gupta A, Popovic ZB, Yong SL, et al. Prevention of cardiac hypertrophy and heart failure by silencing of NF-kappaB. J Mol Biol. 2008 Jan 18;375(3):637–49 doi:10.1016/j.jmb.2007.10.006 PubMed PMID: 18037434; PubMed Central PMCID: PMC2277468.
- Abdellatif M, Dayan S, Fabi SG. Teachings from COVID-19 and aging-An oxidative process. J Cosmet Dermatol. 2020 Dec;19(12):3171–6. doi:10.1111/jocd.13751 PubMed PMID: 32997887; PubMed Central PMCID: PMC7536979.
- Yan M, Sun S, Xu K, Huang X, Dou L, Pang J, et al. Cardiac Aging: From Basic Research to Therapeutics. Oxid Med Cell Longev. 2021;2021:9570325. doi:10.1155/2021/9570325 PubMed PMID: 33777324; PubMed Central PMCID: PMC7969106.
- Furman D, Chang J, Lartigue L, Bolen CR, Haddad F, Gaudilliere B, et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat Med. 2017 Feb;23(2):174–84. doi:10.1038/nm.4267
- Gude NA, Broughton KM, Firouzi F, Sussman MA. Cardiac ageing: extrinsic and intrinsic factors in cellular renewal and senescence. Nat Rev Cardiol. 2018 Sep;15(9):523–42. doi:10.1038/s41569-018-0061-5
- Barcena ML, Aslam M, Pozdniakova S, Norman K, Ladilov Y. Cardiovascular Inflammaging: Mechanisms and Translational Aspects. Cells. 2022 Mar 16;11(6):1010. doi:10.3390/cells11061010
- Palaniappan LP, Allen NB, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, et al. 2026 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association. Circulation. 2026 Mar 3;153(9). doi:10.1161/CIR.0000000000001412
- Kashou AH, Adedinsewo DA, Siontis KC, Noseworthy PA. Artificial Intelligence-Enabled ECG: Physiologic and Pathophysiologic Insights and Implications. Compr Physiol. 2022 Jun 29;12(3):3417–24. doi:10.1002/cphy.c210001 PubMed PMID: 35766831; PubMed Central PMCID: PMC9795459.
- Baek YS, Lee DH, Jo Y, Lee SC, Choi W, Kim DH. Artificial intelligence-estimated biological heart age using a 12-lead electrocardiogram predicts mortality and cardiovascular outcomes. Front Cardiovasc Med. 2023 Apr 13;10:1137892. doi:10.3389/fcvm.2023.1137892
- Lima EM, Ribeiro AH, Paixão GMM, Ribeiro MH, Pinto-Filho MM, Gomes PR, et al. Deep neural network-estimated electrocardiographic age as a mortality predictor. Nat Commun. 2021 Aug 25;12(1):5117. doi:10.1038/s41467-021-25351-7
- Ladejobi AO, Medina-Inojosa JR, Shelly Cohen M, Attia ZI, Scott CG, LeBrasseur NK, et al. The 12-lead electrocardiogram as a biomarker of biological age. Eur Heart J - Digit Health. 2021 Sep 30;2(3):379–89. doi:10.1093/ehjdh/ztab043
- Evans S, Howson SA, Booth AEC, Shahmohamadi E, Lim M, Bacchi S, et al. Artificial intelligence electrocardiogram-predicted biological age gap and mortality: Capturing dynamic risk with multiple electrocardiograms. Heart Rhythm. 2025 May;S1547527125024324. doi:10.1016/j.hrthm.2025.05.009
- Luxenburger H, Thimme R, Hofmann M. T cell adaptation in chronic infections and tumors. Cell Mol Immunol. 2026 Mar 30;23(5):440–56. doi:10.1038/s41423-026-01405-y
- Li F, Liu H, Zhang D, Ma Y, Zhu B. Metabolic plasticity and regulation of T cell exhaustion. Immunology. 2022 Dec;167(4):482–94. doi:10.1111/imm.13575
- Huang X, Fan W, Sun J, Yang J, Zhang Y, Wang Q, et al. SARS-CoV-2 induces cardiomyocyte apoptosis and inflammation but can be ameliorated by ACE inhibitor Captopril. Antiviral Res. 2023 Jul;215:105636 doi:10.1016/j.antiviral.2023.105636 PubMed PMID: 37207821; PubMed Central PMCID: PMC10191704.
- Chung MK, Zidar DA, Bristow MR, Cameron SJ, Chan T, Harding CV, et al. COVID-19 and Cardiovascular Disease: From Bench to Bedside. Circ Res. 2021 Apr 16;128(8):1214–36. doi:10.1161/CIRCRESAHA.121.317997 PubMed PMID: 33856918; PubMed Central PMCID: PMC8048382.
- Shang C, Liu Z, Zhu Y, Lu J, Ge C, Zhang C, et al. SARS-CoV-2 Causes Mitochondrial Dysfunction and Mitophagy Impairment. Front Microbiol. 2021;12:780768. doi:10.3389/fmicb.2021.780768 PubMed PMID: 35069483; PubMed Central PMCID: PMC8770829.
- Molnar T, Lehoczki A, Fekete M, Varnai R, Zavori L, Erdo-Bonyar S, et al. Mitochondrial dysfunction in long COVID: mechanisms, consequences, and potential therapeutic approaches. GeroScience. 2024 Oct;46(5):5267–86. doi:10.1007/s11357-024-01165-5 PubMed PMID: 38668888; PubMed Central PMCID: PMC11336094.
- Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020 Apr 16;181(2):281-292.e6. doi:10.1016/j.cell.2020.02.058 PubMed PMID: 32155444; PubMed Central PMCID: PMC7102599.
- Chung MK, Zidar DA, Bristow MR, Cameron SJ, Chan T, Harding CV, et al. COVID-19 and Cardiovascular Disease: From Bench to Bedside. Circ Res. 2021 Apr 16;128(8):1214–36. doi:10.1161/CIRCRESAHA.121.317997 PubMed PMID: 33856918; PubMed Central PMCID: PMC8048382.
- Oudit GY, Wang K, Viveiros A, Kellner MJ, Penninger JM. Angiotensin-converting enzyme 2-at the heart of the COVID-19 pandemic. Cell. 2023 Mar 2;186(5):906–22. doi:10.1016/j.cell.2023.01.039 PubMed PMID: 36787743; PubMed Central PMCID: PMC9892333.
- Oudit GY, Kassiri Z, Jiang C, Liu PP, Poutanen SM, Penninger JM, et al. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest. 2009 Jul;39(7):618–25. doi:10.1111/j.1365-2362.2009.02153.x PubMed PMID: 19453650; PubMed Central PMCID: PMC7163766.
- Sampson AK, Moritz KM, Denton KM. Postnatal Ontogeny of Angiotensin Receptors and ACE2 in Male and Female Rats. Gend Med. 2012 Feb;9(1):21–32. doi:10.1016/j.genm.2011.12.003
- Bártová E, Legartová S, Krejčí J, Arcidiacono OA. Cell differentiation and aging accompanied by depletion of the ACE2 protein. Aging. 2020 Nov 30;22495–508. doi:10.18632/aging.202221
- Shafqat A, Masters MC, Tripathi U, Tchkonia T, Kirkland JL, Hashmi SK. Long COVID as a disease of accelerated biological aging: An opportunity to translate geroscience interventions. Ageing Res Rev. 2024 Aug;99:102400. doi:10.1016/j.arr.2024.102400
- Zazzara MB, Bellieni A, Calvani R, Coelho-Junior HJ, Picca A, Marzetti E. Inflammaging at the Time of COVID-19. Clin Geriatr Med. 2022 Aug;38(3):473–81. doi:10.1016/j.cger.2022.03.003 PubMed PMID: 35868667; PubMed Central PMCID: PMC8934712.
- Tsampasian V, Bäck M, Bernardi M, Cavarretta E, Dębski M, Gati S, et al. Cardiovascular disease as part of Long COVID: a systematic review. Eur J Prev Cardiol. 2025 Apr 22;32(6):485–98. doi:10.1093/eurjpc/zwae070 PubMed PMID: 38381595.
- Files JK, Sarkar S, Fram TR, Boppana S, Sterrett S, Qin K, et al. Duration of post-COVID-19 symptoms is associated with sustained SARS-CoV-2-specific immune responses. JCI Insight. 2021 Aug 9;6(15):e151544. doi:10.1172/jci.insight.151544 PubMed PMID: 34143754; PubMed Central PMCID: PMC8410022.
- Dobrowolska K, Zarębska-Michaluk D, Poniedziałek B, Jaroszewicz J, Flisiak R, Rzymski P. Overview of autoantibodies in COVID-19 convalescents. J Med Virol. 2023 Jun;95(6):e28864. doi:10.1002/jmv.28864 PubMed PMID: 37310140.
- Undas A, Musiał J, Ząbczyk M. Antiphospholipid antibodies and atherosclerotic vascular disease: recent advances. Rheumatol Int. 2025 Nov 29;45(12):279. doi:10.1007/s00296-025-06050-8 PubMed PMID: 41317185; PubMed Central PMCID: PMC12664860.
- Ryu JK, Sozmen EG, Dixit K, Montano M, Matsui Y, Liu Y, et al. SARS-CoV-2 spike protein induces abnormal inflammatory blood clots neutralized by fibrin immunotherapy. BioRxiv Prepr Serv Biol. 2021 Oct 13;2021.10.12.464152. doi:10.1101/2021.10.12.464152 PubMed PMID: 34671772; PubMed Central PMCID: PMC8528086.
- Patterson BK, Francisco EB, Yogendra R, Long E, Pise A, Rodrigues H, et al. Persistence of SARS CoV-2 S1 Protein in CD16+ Monocytes in Post-Acute Sequelae of COVID-19 (PASC) up to 15 Months Post-Infection. Front Immunol. 2021;12:746021. doi:10.3389/fimmu.2021.746021 PubMed PMID: 35082777; PubMed Central PMCID: PMC8784688.
- Shang C, Liu Z, Zhu Y, Lu J, Ge C, Zhang C, et al. SARS-CoV-2 Causes Mitochondrial Dysfunction and Mitophagy Impairment. Front Microbiol. 2021;12:780768. doi:10.3389/fmicb.2021.780768 PubMed PMID: 35069483; PubMed Central PMCID: PMC8770829.
- Huang X, Fan W, Sun J, Yang J, Zhang Y, Wang Q, et al. SARS-CoV-2 induces cardiomyocyte apoptosis and inflammation but can be ameliorated by ACE inhibitor Captopril. Antiviral Res. 2023 Jul;215:105636. doi:10.1016/j.antiviral.2023.105636 PubMed PMID: 37207821; PubMed Central PMCID: PMC10191704.
- Shen Y, Chen M, Gu W, Wan J, Cheng Z, Shen K, et al. The molecular mechanism of cardiac injury in SARS-CoV-2 infection: Focus on mitochondrial dysfunction. J Infect Public Health. 2023 May;16(5):746–53. doi:10.1016/j.jiph.2023.03.015 PubMed PMID: 36958170; PubMed Central PMCID: PMC10019919.
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998 Aug 28;281(5381):1309–12. doi:10.1126/science.281.5381.1309 PubMed PMID: 9721092.
- Xie S, Xu SC, Deng W, Tang Q. Metabolic landscape in cardiac aging: insights into molecular biology and therapeutic implications. Signal Transduct Target Ther. 2023 Mar 14;8(1):114. doi:10.1038/s41392-023-01378-8 PubMed PMID: 36918543; PubMed Central PMCID: PMC10015017.
- Raman B, Bluemke DA, Lüscher TF, Neubauer S. Long COVID: post-acute sequelae of COVID-19 with a cardiovascular focus. Eur Heart J. 2022 Mar 14;43(11):1157–72 doi:10.1093/eurheartj/ehac031 PubMed PMID: 35176758; PubMed Central PMCID: PMC8903393.
- Qiu J, Söderlund-Venermo M, Young NS. Human Parvoviruses. Clin Microbiol Rev. 2017 Jan;30(1):43–113. doi:10.1128/CMR.00040-16 PubMed PMID: 27806994; PubMed Central PMCID: PMC5217800.
- Schultheiss HP, Baumeier C, Pietsch H, Bock CT, Poller W, Escher F. Cardiovascular consequences of viral infections: from COVID to other viral diseases. Cardiovasc Res. 2021 Nov 22;117(13):2610–23. doi:10.1093/cvr/cvab315 PubMed PMID: 34609508; PubMed Central PMCID: PMC8500164.
- Seals DR, Jablonski KL, Donato AJ. Aging and vascular endothelial function in humans. Clin Sci Lond Engl 1979 2011 May;120(9):357–75. doi:10.1042/CS20100476 PubMed PMID: 21244363; PubMed Central PMCID: PMC3482987.
- Schmidt-Lucke C, Zobel T, Schrepfer S, Kuhl U, Wang D, Klingel K, et al. Impaired Endothelial Regeneration Through Human Parvovirus B19-Infected Circulating Angiogenic Cells in Patients With Cardiomyopathy. J Infect Dis. 2015 Oct 1;212(7):1070–81. doi:10.1093/infdis/jiv178 PubMed PMID: 25805750.
- Van Linthout S, Elsanhoury A, Klein O, Sosnowski M, Miteva K, Lassner D, et al. Telbivudine in chronic lymphocytic myocarditis and human parvovirus B19 transcriptional activity. ESC Heart Fail. 2018 Oct;5(5):818–29. doi:10.1002/ehf2.12341 PubMed PMID: 30099854; PubMed Central PMCID: PMC6165949.
- Yue Y, Meng K, Pu Y, Zhang X. Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res Clin Pract. 2017 Nov;133:124–30. doi:10.1016/j.diabres.2017.08.018 PubMed PMID: 28934669.
- Shimojo N, Hashizume R, Kanayama K, Hara M, Suzuki Y, Nishioka T, et al. Tenascin-C may accelerate cardiac fibrosis by activating macrophages via the integrin αVβ3/nuclear factor-κB/interleukin-6 axis. Hypertens Dallas Tex 1979 2015 Oct;66(4):757–66. doi:10.1161/HYPERTENSIONAHA.115.06004 PubMed PMID: 26238448.
- Rinkūnaitė I, Šimoliūnas E, Bironaitė D, Rutkienė R, Bukelskienė V, Meškys R, et al. The Effect of a Unique Region of Parvovirus B19 Capsid Protein VP1 on Endothelial Cells. Biomolecules. 2021 Apr 19;11(4):606. doi:10.3390/biom11040606 PubMed PMID: 33921883; PubMed Central PMCID: PMC8073096.
- Totoń-Żurańska J, Mikolajczyk TP, Saju B, Guzik TJ. Vascular remodelling in cardiovascular diseases: hypertension, oxidation, and inflammation. Clin Sci Lond Engl 1979. 2024 Jul 3;138(13):817–50 doi:10.1042/CS20220797 PubMed PMID: 38920058.
- Escher F, Aleshcheva G, Pietsch H, Baumeier C, Gross UM, Schrage BN, et al. Transcriptional Active Parvovirus B19 Infection Predicts Adverse Long-Term Outcome in Patients with Non-Ischemic Cardiomyopathy. Biomedicines. 2021 Dec 14;9(12):1898. doi:10.3390/biomedicines9121898 PubMed PMID: 34944716; PubMed Central PMCID: PMC8698988.
- Kuhl U, Lassner D, Dorner A, Rohde M, Escher F, Seeberg B, et al. A distinct subgroup of cardiomyopathy patients characterized by transcriptionally active cardiotropic erythrovirus and altered cardiac gene expression. Basic Res Cardiol. 2013 Sep;108(5):372 doi:10.1007/s00395-013-0372-y PubMed PMID: 23934091.
- Tzang CC, Chi LY, Lee CY, Chang ZY, Luo CA, Chen YH, et al. Clinical implications of human Parvovirus B19 infection on autoimmunity and autoimmune diseases. Int Immunopharmacol. 2025 Feb 6;147:113960. doi:10.1016/j.intimp.2024.113960 PubMed PMID: 39746271.
- Tzang BS, Lin TM, Tsai CC, Hsu JD, Yang LC, Hsu TC. Increased cardiac injury in NZB/W F1 mice received antibody against human parvovirus B19 VP1 unique region protein. Mol Immunol. 2011 Jul;48(12–13):1518–24. doi:10.1016/j.molimm.2011.04.013 PubMed PMID: 21555155.
- Chen DY, Tzang BS, Chen YM, Lan JL, Tsai CC, Hsu TC. The association of anti-parvovirus B19-VP1 unique region antibodies with antiphospholipid antibodies in patients with antiphospholipid syndrome. Clin Chim Acta. 2010 Aug;411(15–16):1084–9. doi:10.1016/j.cca.2010.04.004
- Chou TNK, Hsu TC, Chen RM, Lin LI, Tsay GJ. Parvovirus B19 infection associated with the production of antineutrophil cytoplasmic antibody (ANCA) and anticardiolipin antibody (aCL). Lupus. 2000 Sep;9(7):551–4. doi:10.1177/096120330000900714
- Elsanhoury A, Kühl U, Stautner B, Klein O, Krannich A, Morris D, et al. The Spontaneous Course of Human Herpesvirus 6 DNA-Associated Myocarditis and the Effect of Immunosuppressive Intervention. Viruses. 2022 Jan 31;14(2):299. doi:10.3390/v14020299 PubMed PMID: 35215893; PubMed Central PMCID: PMC8879301.
- Schreiner P, Harrer T, Scheibenbogen C, Lamer S, Schlosser A, Naviaux RK, et al. Human Herpesvirus-6 Reactivation, Mitochondrial Fragmentation, and the Coordination of Antiviral and Metabolic Phenotypes in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. ImmunoHorizons. 2020 Apr 23;4(4):201–15. doi:10.4049/immunohorizons.2000006 PubMed PMID: 32327453.
- Wu Z, Jia J, Xu X, Xu M, Peng G, Ma J, et al. Human herpesvirus 6A promotes glycolysis in infected T cells by activation of mTOR signaling. PLoS Pathog. 2020 Jun;16(6):e1008568. doi:10.1371/journal.ppat.1008568 PubMed PMID: 32516328; PubMed Central PMCID: PMC7282626.
- Lesnefsky EJ, Chen Q, Hoppel CL. Mitochondrial Metabolism in Aging Heart. Circ Res. 2016 May 13;118(10):1593–611. doi:10.1161/CIRCRESAHA.116.307505
- Golob S, Nazeer H, Kadosh B, Goldberg R, Narula N, Moazami N, et al. HHV-6 Myocarditis Progressing to Ventricular Standstill Requiring Cardiac Transplant. JACC Case Rep. 2023 Jul;17:101896. doi:10.1016/j.jaccas.2023.101896
- Yamada A, Takeichi T, Kiryu K, Takashino S, Yoshida M, Kitamura O. Fatal human herpes virus 6B myocarditis: Postmortem diagnosis of HHV-6B based on CD134+ T-cell tropism. Leg Med. 2022 Feb;54:102007. doi:10.1016/j.legalmed.2021.102007
- Kühl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W, et al. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation. 2005 Sep 27;112(13):1965–70. doi:10.1161/CIRCULATIONAHA.105.548156 PubMed PMID: 16172268.
- Meier UC, Cipian RC, Karimi A, Ramasamy R, Middeldorp JM. Cumulative Roles for Epstein-Barr Virus, Human Endogenous Retroviruses, and Human Herpes Virus-6 in Driving an Inflammatory Cascade Underlying MS Pathogenesis. Front Immunol. 2021;12:757302. doi:10.3389/fimmu.2021.757302 PubMed PMID: 34790199; PubMed Central PMCID: PMC8592026.
- Marrie RA, Fisk JD, Fitzgerald K, Kowalec K, Maxwell C, Rotstein D, et al. Etiology, effects and management of comorbidities in multiple sclerosis: recent advances. Front Immunol. 2023;14:1197195. doi:10.3389/fimmu.2023.1197195 PubMed PMID: 37325663; PubMed Central PMCID: PMC10266935.
- Granato M, Gilardini Montani MS, Zompetta C, Santarelli R, Gonnella R, Romeo MA, et al. Quercetin Interrupts the Positive Feedback Loop Between STAT3 and IL-6, Promotes Autophagy, and Reduces ROS, Preventing EBV-Driven B Cell Immortalization. Biomolecules. 2019 Sep 12;9(9):482. doi:10.3390/biom9090482 PubMed PMID: 31547402; PubMed Central PMCID: PMC6769872.
- Zheng Q, Wang X. Autophagy and the ubiquitin-proteasome system in cardiac dysfunction. Panminerva Med. 2010 Mar;52(1):9–25. PubMed PMID: 20228723; PubMed Central PMCID: PMC2840262.
- Yi L, Hu Y, Wu Z, Li Y, Kong M, Kang Z, et al. TFRC upregulation promotes ferroptosis in CVB3 infection via nucleus recruitment of Sp1. Cell Death Dis. 2022 Jul 11;13(7):592. doi:10.1038/s41419-022-05027-w PubMed PMID: 35821227; PubMed Central PMCID: PMC9276735.
- Liu X, Li M, Chen Z, Yu Y, Shi H, Yu Y, et al. Mitochondrial calpain-1 activates NLRP3 inflammasome by cleaving ATP5A1 and inducing mitochondrial ROS in CVB3-induced myocarditis. Basic Res Cardiol. 2022 Aug 23;117(1):40. doi:10.1007/s00395-022-00948-1 PubMed PMID: 35997820; PubMed Central PMCID: PMC9399059.
- Oh SJ, Lim BK, Yun J, Shin OS. CVB3-Mediated Mitophagy Plays an Important Role in Viral Replication via Abrogation of Interferon Pathways. Front Cell Infect Microbiol. 2021;11:704494. doi:10.3389/fcimb.2021.704494 PubMed PMID: 34295842; PubMed Central PMCID: PMC8292102.
- Bredow C, Thery F, Wirth EK, Ochs S, Kespohl M, Kleinau G, et al. ISG15 blocks cardiac glycolysis and ensures sufficient mitochondrial energy production during Coxsackievirus B3 infection. Cardiovasc Res. 2024 May 7;120(6):644–57. doi:10.1093/cvr/cvae026 PubMed PMID: 38309955; PubMed Central PMCID: PMC11074791.
- Fairweather D, Frisancho-Kiss S, Rose NR. Viruses as adjuvants for autoimmunity: evidence from Coxsackievirus-induced myocarditis. Rev Med Virol. 2005;15(1):17–27. doi:10.1002/rmv.445 PubMed PMID: 15386590.
- Shi J, Wong J, Piesik P, Fung G, Zhang J, Jagdeo J, et al. Cleavage of sequestosome 1/p62 by an enteroviral protease results in disrupted selective autophagy and impaired NFKB signaling. Autophagy. 2013 Oct;9(10):1591–603. doi:10.4161/auto.26059 PubMed PMID: 23989536.
- Shi J, Fung G, Piesik P, Zhang J, Luo H. Dominant-negative function of the C-terminal fragments of NBR1 and SQSTM1 generated during enteroviral infection. Cell Death Differ. 2014 Sep;21(9):1432–41. doi:10.1038/cdd.2014.58 PubMed PMID: 24769734; PubMed Central PMCID: PMC4131175.
- Mohamud Y, Qu J, Xue YC, Liu H, Deng H, Luo H. CALCOCO2/NDP52 and SQSTM1/p62 differentially regulate coxsackievirus B3 propagation. Cell Death Differ. 2019 Jun;26(6):1062–76. doi:10.1038/s41418-018-0185-5 PubMed PMID: 30154446; PubMed Central PMCID: PMC6748094.
- Sanbe A, Osinska H, Saffitz JE, Glabe CG, Kayed R, Maloyan A, et al. Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci U S A. 2004 Jul 6;101(27):10132–6. doi:10.1073/pnas.0401900101 PubMed PMID: 15220483; PubMed Central PMCID: PMC454177.
- Qiu Y, Xu J, Chen Y, Wu Y, Lin YN, Liu W, et al. Parkin plays a crucial role in acute viral myocarditis by regulating mitophagy activity. Theranostics. 2024;14(13):5303–15. doi:10.7150/thno.97675 PubMed PMID: 39267792; PubMed Central PMCID: PMC11388078.
- Sin J, Mangale V, Thienphrapa W, Gottlieb RA, Feuer R. Recent progress in understanding coxsackievirus replication, dissemination, and pathogenesis. Virology. 2015 Oct;484:288–304. doi:10.1016/j.virol.2015.06.006 PubMed PMID: 26142496; PubMed Central PMCID: PMC4567421.
- Yip F, Lai B, Yang D. Role of Coxsackievirus B3-Induced Immune Responses in the Transition from Myocarditis to Dilated Cardiomyopathy and Heart Failure. Int J Mol Sci. 2023 Apr 23;24(9):7717. doi:10.3390/ijms24097717 PubMed PMID: 37175422; PubMed Central PMCID: PMC10178405.
- Sin J, McIntyre L, Stotland A, Feuer R, Gottlieb RA. Coxsackievirus B Escapes the Infected Cell in Ejected Mitophagosomes. J Virol. 2017 Dec 15;91(24):e01347-17. doi:10.1128/JVI.01347-17 PubMed PMID: 28978702; PubMed Central PMCID: PMC5709598.
- Mohamud Y, Bahreyni A, Hwang SW, Lin JC, Wang ZC, Zhang J, et al. Mitochondrial injury and complement dysregulation are drivers of pathological inflammation in viral myocarditis. J Virol. 2025 Feb 25;99(2):e0180424. doi:10.1128/jvi.01804-24 PubMed PMID: 39846741; PubMed Central PMCID: PMC11852726.
- Sengupta PP, Jacob SS, Suresh KP, Rajamani S, Maharana SM. Exploring global trends in human toxoplasmosis seroprevalence by meta-analysis. Exp Parasitol. 2025 Aug;275:108971. doi:10.1016/j.exppara.2025.108971 PubMed PMID: 40562157.
- Alhusseiny SM, Saleh NE, El-Zayady WM, Hussein MS, El-Beshbishi SN. Association between Toxoplasma gondii infection and coronary atherosclerosis. Trans R Soc Trop Med Hyg. 2021 Oct 1;115(10):1190–7. doi:10.1093/trstmh/trab018 PubMed PMID: 33596320.
- Sana M, Rashid M, Rashid I, Akbar H, Gomez-Marin JE, Dimier-Poisson I. Immune response against toxoplasmosis-some recent updates RH: Toxoplasma gondii immune response. Int J Immunopathol Pharmacol. 2022;36:3946320221078436. doi:10.1177/03946320221078436 PubMed PMID: 35227108; PubMed Central PMCID: PMC8891885.
- Gov L, Schneider CA, Lima TS, Pandori W, Lodoen MB. NLRP3 and Potassium Efflux Drive Rapid IL-1β Release from Primary Human Monocytes during Toxoplasma gondii Infection. J Immunol. 2017 Oct 15;199(8):2855–64. doi:10.4049/jimmunol.1700245
- Moreira-Souza ACA, Almeida-da-Silva CLC, Rangel TP, Rocha GDC, Bellio M, Zamboni DS, et al. The P2X7 Receptor Mediates Toxoplasma gondii Control in Macrophages through Canonical NLRP3 Inflammasome Activation and Reactive Oxygen Species Production. Front Immunol. 2017 Oct 12;8:1257. doi:10.3389/fimmu.2017.01257
- Sena CM, Leandro A, Azul L, Seiça R, Perry G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front Physiol. 2018;9:1668 doi:10.3389/fphys.2018.01668 PubMed PMID: 30564132; PubMed Central PMCID: PMC6288353.
- Egorov AI, Converse RR, Griffin SM, Styles JN, Sams E, Hudgens E, et al. Latent Toxoplasma gondii infections are associated with elevated biomarkers of inflammation and vascular injury. BMC Infect Dis. 2021 Feb 18;21(1):188. doi:10.1186/s12879-021-05882-6 PubMed PMID: 33602170; PubMed Central PMCID: PMC7890825.
- Al-Kuraishi AH, Al-Windy SD, Al-Kuraishy HM, Al-Gareeb AI. Endothelial dysfunction in acute acquired toxoplasmosis. Trop Parasitol. 2020;10(1):29–33. doi:10.4103/tp.TP_26_19 PubMed PMID: 32775289; PubMed Central PMCID: PMC7365500.
- Hoover EM, Schneider CA, Crouzet C, Lima TS, Velez DXF, Tran CJ, et al. Infection with Toxoplasma gondii triggers coagulation at the blood-brain barrier and a reduction in cerebral blood flow. J Neuroinflammation. 2025 Jan 8;22(1):3. doi:10.1186/s12974-024-03330-1
- Velásquez ZD, Rojas-Baron L, Conejeros I, Hermosilla C, Taubert A. Toxoplasma gondii infection induces early host cell cycle arrest and DNA damage in primary human host cells by a MYR1-dependent mechanism. Commun Biol. 2024 Dec 16;7(1):1637. doi:10.1038/s42003-024-07374-0 PubMed PMID: 39681694; PubMed Central PMCID: PMC11649780.
- Babekir A, Mostafa S, Obeng-Gyasi E. The Association of Toxoplasma gondii IgG and Cardiovascular Biomarkers. Int J Environ Res Public Health. 2021 May 5;18(9):4908. doi:10.3390/ijerph18094908 PubMed PMID: 34062965; PubMed Central PMCID: PMC8125424.
- Alvarado-Esquivel C, Salcedo-Jaquez M, Sanchez-Anguiano LF, Hernandez-Tinoco J, Rabago-Sanchez E, Beristain-Garcia I, et al. Association Between Toxoplasma gondii Exposure and Heart Disease: A Case-Control Study. J Clin Med Res. 2016 May;8(5):402–9. doi:10.14740/jocmr2525w PubMed PMID: 27081427; PubMed Central PMCID: PMC4817581.
- Dragomir A, Lupu MA, Lighezan R, Paduraru AA, Olariu TR. Toxoplasma gondii Infection in Patients with Cardiovascular Diseases from Western Romania: A Case–Control Study. Life. 2023 Jul 17;13(7):1575. doi:10.3390/life13071575
- Huang L, You X, Lu Z, Zhou X, He L, Zou C, et al. Toxoplasma Gondii infection and cardiovascular mortality: sex-specific differences in a United States population-based cohort study. BMC Infect Dis. 2024 Sep 27;24(1):1029. doi:10.1186/s12879-024-09934-5 PubMed PMID: 39333926; PubMed Central PMCID: PMC11428453.
- Bern C. Chagas’ Disease. N Engl J Med. 2015 Jul 30;373(5):456–66. doi:10.1056/NEJMra1410150 PubMed PMID: 26222561.
- American trypanosomiasis. In: Handbook of Clinical Neurology [Internet]. Elsevier; 2013 [cited 2025 Dec 17]. p. 103–23. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780444534903000078 doi:10.1016/B978-0-444-53490-3.00007-8
- Beatty NL, Hamer GL, Moreno-Peniche B, Mayes B, Hamer SA. Chagas Disease, an Endemic Disease in the United States. Emerg Infect Dis. 2025 Sep;31(9). doi:10.3201/eid3109.241700
- Kirchhoff LV. Epidemiology of American trypanosomiasis (Chagas disease). Adv Parasitol. 2011;75:1–18. doi:10.1016/B978-0-12-385863-4.00001-0 PubMed PMID: 21820549.
- Rassi A, Rassi A, Marcondes de Rezende J. American trypanosomiasis (Chagas disease). Infect Dis Clin North Am. 2012 Jun;26(2):275–91. doi:10.1016/j.idc.2012.03.002 PubMed PMID: 22632639.
- Montalvo-Ocotoxtle IG, Rojas-Velasco G, Rodríguez-Morales O, Arce-Fonseca M, Baeza-Herrera LA, Arzate-Ramírez A, et al. Chagas Heart Disease: Beyond a Single Complication, from Asymptomatic Disease to Heart Failure. J Clin Med. 2022 Dec 7;11(24):7262. doi:10.3390/jcm11247262 PubMed PMID: 36555880; PubMed Central PMCID: PMC9784121.
- Frade AF, Guérin H, Nunes JPS, Silva LFSE, Roda VM de P, Madeira RP, et al. Cardiac and Digestive Forms of Chagas Disease: An Update on Pathogenesis, Genetics, and Therapeutic Targets. Mediators Inflamm. 2025;2025:8862004. doi:10.1155/mi/8862004 PubMed PMID: 40297326; PubMed Central PMCID: PMC12037249.
- Brito BOF, Lima EM, Soliman EZ, Silva EF, Lima-Costa MF, Ribeiro ALP. The evolution of electrocardiographic abnormalities in the elderly with Chagas disease during 14 years of follow-up: The Bambui Cohort Study of Aging. PLoS Negl Trop Dis. 2023 Jun;17(6):e0011419. doi:10.1371/journal.pntd.0011419 PubMed PMID: 37285382; PubMed Central PMCID: PMC10281574.
- Cutshaw MK, Sciaudone M, Bowman NM. Risk Factors for Progression to Chronic Chagas Cardiomyopathy: A Systematic Review and Meta-Analysis. Am J Trop Med Hyg. 2023 Apr 5;108(4):791–800. doi:10.4269/ajtmh.22-0630 PubMed PMID: 36848894; PubMed Central PMCID: PMC10076993.
- Nunes JPS, Roda VM de P, Andrieux P, Kalil J, Chevillard C, Cunha-Neto E. Inflammation and mitochondria in the pathogenesis of chronic Chagas disease cardiomyopathy. Exp Biol Med Maywood NJ. 2023 Nov;248(22):2062–71 doi:10.1177/15353702231220658 PubMed PMID: 38235691; PubMed Central PMCID: PMC10800136.
- Teixeira PC, Ducret A, Langen H, Nogoceke E, Santos RHB, Silva Nunes JP, et al. Impairment of Multiple Mitochondrial Energy Metabolism Pathways in the Heart of Chagas Disease Cardiomyopathy Patients. Front Immunol. 2021;12:755782 doi:10.3389/fimmu.2021.755782 PubMed PMID: 34867990; PubMed Central PMCID: PMC8633876.
- Rodrigues MM, Oliveira AC, Bellio M. The Immune Response to Trypanosoma cruzi: Role of Toll-Like Receptors and Perspectives for Vaccine Development. J Parasitol Res. 2012;2012:507874. doi:10.1155/2012/507874 PubMed PMID: 22496959; PubMed Central PMCID: PMC3306967.
- da Matta Guedes PM, Gutierrez FRS, Maia FL, Milanezi CM, Silva GK, Pavanelli WR, et al. IL-17 produced during Trypanosoma cruzi infection plays a central role in regulating parasite-induced myocarditis. PLoS Negl Trop Dis. 2010 Feb 16;4(2):e604. doi:10.1371/journal.pntd.0000604 PubMed PMID: 20169058; PubMed Central PMCID: PMC2821906.
- Lopez M, Tanowitz HB, Garg NJ. Pathogenesis of Chronic Chagas Disease: Macrophages, Mitochondria, and Oxidative Stress. Curr Clin Microbiol Rep. 2018 Mar;5(1):45–54. PubMed PMID: 29868332; PubMed Central PMCID: PMC5983038.
- Paiva CN, Medei E, Bozza MT. ROS and Trypanosoma cruzi: Fuel to infection, poison to the heart. PLoS Pathog. 2018 Apr;14(4):e1006928. doi:10.1371/journal.ppat.1006928 PubMed PMID: 29672619; PubMed Central PMCID: PMC5908069.
- Nogueira SS, Souza MA, Santos EC, Caldas IS, Gonçalves RV, Novaes RD. Oxidative stress, cardiomyocytes premature senescence and contractile dysfunction in in vitro and in vivo experimental models of Chagas disease. Acta Trop. 2023 Aug;244:106950. doi:10.1016/j.actatropica.2023.106950 PubMed PMID: 37211152.
- Sousa GR, Gomes JAS, Fares RCG, Damásio MP de S, Chaves AT, Ferreira KS, et al. Plasma cytokine expression is associated with cardiac morbidity in chagas disease. PloS One. 2014;9(3):e87082. doi:10.1371/journal.pone.0087082 PubMed PMID: 24603474; PubMed Central PMCID: PMC3945957.
- Souza-Silva TG, Neves EGA, Teixeira-Carvalho A, Figueiredo AB, Morais KLP, Apostólico J, et al. Self and parasite-derived peptides selected upon DERAA-bearing HLA-DRB1 alleles activate CD4+ T cells from Chagas cardiomyopathy patients and are associated with ventricular dysfunction. Front Immunol. 2025;16:1527115. doi:10.3389/fimmu.2025.1527115 PubMed PMID: 40391216; PubMed Central PMCID: PMC12086171.
- Sarocchi M, Gayraud JC, Dajoux R, Martin-Laval J [Letter: Relative pH of the cervical mucus and sperm in sterility].. Nouv Presse Med. 1975 May 24;4(21):1595–6. PubMed PMID: 2903.
- Poncini CV, Benatar AF, Gomez KA, Rabinovich GA. Galectins in Chagas Disease: A Missing Link Between Trypanosoma cruzi Infection, Inflammation, and Tissue Damage. Front Microbiol. 2021;12:794765. doi:10.3389/fmicb.2021.794765 PubMed PMID: 35046919; PubMed Central PMCID: PMC8762303.
- Benatar AF, García GA, Bua J, Cerliani JP, Postan M, Tasso LM, et al. Galectin-1 Prevents Infection and Damage Induced by Trypanosoma cruzi on Cardiac Cells. PLoS Negl Trop Dis. 2015;9(10):e0004148. doi:10.1371/journal.pntd.0004148 PubMed PMID: 26451839; PubMed Central PMCID: PMC4599936.
- Poncini CV, Ilarregui JM, Batalla EI, Engels S, Cerliani JP, Cucher MA, et al. Trypanosoma cruzi Infection Imparts a Regulatory Program in Dendritic Cells and T Cells via Galectin-1-Dependent Mechanisms. J Immunol Baltim Md 1950. 2015 Oct 1;195(7):3311–24. doi:10.4049/jimmunol.1403019 PubMed PMID: 26324777.
- Maldonado E, Rojas DA, Urbina F, Solari A. The Oxidative Stress and Chronic Inflammatory Process in Chagas Disease: Role of Exosomes and Contributing Genetic Factors. Oxid Med Cell Longev. 2021;2021:4993452. doi:10.1155/2021/4993452 PubMed PMID: 34976301; PubMed Central PMCID: PMC8718323.
- Gupta S, Bhatia V, Wen J jun, Wu Y, Huang MH, Garg NJ. Trypanosoma cruzi infection disturbs mitochondrial membrane potential and ROS production rate in cardiomyocytes. Free Radic Biol Med. 2009 Nov 15;47(10):1414–21. doi:10.1016/j.freeradbiomed.2009.08.008 PubMed PMID: 19686837; PubMed Central PMCID: PMC2767388.
- Wen JJ, Porter C, Garg NJ. Inhibition of NFE2L2-Antioxidant Response Element Pathway by Mitochondrial Reactive Oxygen Species Contributes to Development of Cardiomyopathy and Left Ventricular Dysfunction in Chagas Disease. Antioxid Redox Signal. 2017 Sep 20;27(9):550–66. doi:10.1089/ars.2016.6831 PubMed PMID: 28132522; PubMed Central PMCID: PMC5567598.
- Nunes JPS, Andrieux P, Brochet P, Almeida RR, Kitano E, Honda AK, et al. Co-Exposure of Cardiomyocytes to IFN-γ and TNF-α Induces Mitochondrial Dysfunction and Nitro-Oxidative Stress: Implications for the Pathogenesis of Chronic Chagas Disease Cardiomyopathy. Front Immunol. 2021;12:755862. doi:10.3389/fimmu.2021.755862 PubMed PMID: 34867992; PubMed Central PMCID: PMC8632642.
- Pozio E. World distribution of Trichinella spp. infections in animals and humans. Vet Parasitol. 2007 Oct;149(1–2):3–21. doi:10.1016/j.vetpar.2007.07.002
- Crisóstomo-Jorquera V, Landaeta-Aqueveque C. The genus Trichinella and its presence in wildlife worldwide: A review. Transbound Emerg Dis. 2022 Sep;69(5):e1269–79. doi:10.1111/tbed.14554 PubMed PMID: 35398980.
- Bang SH, Park JB, Chee HK, Kim JS, Ko SM, Kim WS, et al. Cardiac Parasitic Infection in Trichinellosis Associated with Right Ventricle Outflow Tract Obstruction. Korean J Thorac Cardiovasc Surg. 2014 Apr 10;47(2):145–8. doi:10.5090/kjtcs.2014.47.2.145
- Mohib O, Clevenbergh P, Truyens C, Morissens M, Castro Rodriguez J. Trichinella spiralis -associated myocarditis mimicking acute myocardial infarction. Acta Clin Belg. 2022 Jan 2;77(1):147–52. doi:10.1080/17843286.2020.1790867
- Yan J, Huang S, Lu F. Galectin-Receptor Interactions Regulates Cardiac Pathology Caused by Trichinella spiralis Infection. Front Immunol. 2021;12:639260. doi:10.3389/fimmu.2021.639260 PubMed PMID: 34093526; PubMed Central PMCID: PMC8175896.
- Yan SW, Zhang R, Guo X, Wang BN, Long SR, Liu RD, et al. Trichinella spiralis dipeptidyl peptidase 1 suppressed macrophage cytotoxicity by promoting M2 polarization via the STAT6/PPARγ pathway. Vet Res. 2023 Sep 13;54(1):77. doi:10.1186/s13567-023-01209-2 PubMed PMID: 37705099; PubMed Central PMCID: PMC10500742.
- Sun Q, Huang J, Gu Y, Liu S, Zhu X. Dynamic changes of macrophage activation in mice infected with Trichinella spiralis. Int Immunopharmacol. 2022 Jul;108:108716 doi:10.1016/j.intimp.2022.108716 PubMed PMID: 35344812.
- Deng G, Deng R, Yao J, Liao B, Chen Y, Wu Z, et al. Trichinella spiralis infection changes immune response in mice performed abdominal heterotopic cardiac transplantation and prolongs cardiac allograft survival time. Parasitol Res. 2016 Jan;115(1):407–14. doi:10.1007/s00436-015-4762-y PubMed PMID: 26481486.
- Gehlken C, Suthahar N, Meijers WC, De Boer RA. Galectin-3 in Heart Failure. Heart Fail Clin. 2018 Jan;14(1):75–92. doi:10.1016/j.hfc.2017.08.009
- Ho JE, Liu C, Lyass A, Courchesne P, Pencina MJ, Vasan RS, et al. Galectin-3, a Marker of Cardiac Fibrosis, Predicts Incident Heart Failure in the Community. J Am Coll Cardiol. 2012 Oct;60(14):1249–56. doi:10.1016/j.jacc.2012.04.053
- Chienwichai P, Thiangtrongjit T, Tipthara P, Tarning J, Adisakwattana P, Reamtong O. Untargeted serum metabolomics analysis of Trichinella spiralis-infected mouse. PLoS Negl Trop Dis. 2023 Feb;17(2):e0011119. doi:10.1371/journal.pntd.0011119 PubMed PMID: 36809241; PubMed Central PMCID: PMC9943014.
- Yu H, Yang F, Zhong W, Jiang X, Zhang F, Ji X, et al. Secretory Galectin-3 promotes hepatic steatosis via regulation of the PPARγ/CD36 signaling pathway. Cell Signal. 2021 Aug;84:110043. doi:10.1016/j.cellsig.2021.110043 PubMed PMID: 33991615.
- Sun Z, Zhang L, Li L, Shao C, Liu J, Zhou M, et al. Galectin-3 mediates cardiac remodeling caused by impaired glucose and lipid metabolism through inhibiting two pathways of activating Akt. Am J Physiol Heart Circ Physiol. 2021 Jan 1;320(1):H364–80. doi:10.1152/ajpheart.00523.2020 PubMed PMID: 33275526.
- Liesenborghs L, Meyers S, Lox M, Criel M, Claes J, Peetermans M, et al. Staphylococcus aureus endocarditis: distinct mechanisms of bacterial adhesion to damaged and inflamed heart valves. Eur Heart J. 2019 Oct 14;40(39):3248–59. doi:10.1093/eurheartj/ehz175 PubMed PMID: 30945735; PubMed Central PMCID: PMC7963134.
- Howden BP, Giulieri SG, Wong Fok Lung T, Baines SL, Sharkey LK, Lee JYH, et al. Staphylococcus aureus host interactions and adaptation. Nat Rev Microbiol. 2023 Jun;21(6):380–95. doi:10.1038/s41579-023-00852-y PubMed PMID: 36707725; PubMed Central PMCID: PMC9882747.
- Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013 Jan 17;493(7432):433–6. doi:10.1038/nature11682 PubMed PMID: 23222518; PubMed Central PMCID: PMC3548046.
- Chen H, Zhang J, He Y, Lv Z, Liang Z, Chen J, et al. Exploring the Role of Staphylococcus aureus in Inflammatory Diseases. Toxins. 2022 Jul 6;14(7):464. doi:10.3390/toxins14070464 PubMed PMID: 35878202; PubMed Central PMCID: PMC9318596.
- Cheung GYC, Bae JS, Otto M. Pathogenicity and virulence of Staphylococcus aureus. Virulence. 2021 Dec;12(1):547–69. doi:10.1080/21505594.2021.1878688 PubMed PMID: 33522395; PubMed Central PMCID: PMC7872022.
- Kim JK, Uchiyama S, Gong H, Stream A, Zhang L, Nizet V. Engineered Biomimetic Platelet Membrane-Coated Nanoparticles Block Staphylococcus aureus Cytotoxicity and Protect Against Lethal Systemic Infection. Eng Beijing China. 2021 Aug;7(8):1149–56. doi:10.1016/j.eng.2020.09.013 PubMed PMID: 39449819; PubMed Central PMCID: PMC11501092.
- Zhu Z, Hu Z, Li S, Fang R, Ono HK, Hu DL. Molecular Characteristics and Pathogenicity of Staphylococcus aureus Exotoxins. Int J Mol Sci. 2023 Dec 28;25(1):395. doi:10.3390/ijms25010395 PubMed PMID: 38203566; PubMed Central PMCID: PMC10778951.
- Holland TL, Baddour LM, Bayer AS, Hoen B, Miro JM, Fowler VG. Infective endocarditis. Nat Rev Dis Primer. 2016 Sep 1;2:16059. doi:10.1038/nrdp.2016.59 PubMed PMID: 27582414; PubMed Central PMCID: PMC5240923.
- Ahmad-Mansour N, Loubet P, Pouget C, Dunyach-Remy C, Sotto A, Lavigne JP, et al. Staphylococcus aureus Toxins: An Update on Their Pathogenic Properties and Potential Treatments. Toxins. 2021 Sep 23;13(10):677. doi:10.3390/toxins13100677 PubMed PMID: 34678970; PubMed Central PMCID: PMC8540901.
- Murdoch DR, Corey GR, Hoen B, Miró JM, Fowler VG, Bayer AS, et al. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: the International Collaboration on Endocarditis-Prospective Cohort Study. Arch Intern Med. 2009 Mar 9;169(5):463–73. doi:10.1001/archinternmed.2008.603 PubMed PMID: 19273776; PubMed Central PMCID: PMC3625651.
- Tubiana S, Duval X, Alla F, Selton-Suty C, Tattevin P, Delahaye F, et al. The VIRSTA score, a prediction score to estimate risk of infective endocarditis and determine priority for echocardiography in patients with Staphylococcus aureus bacteremia. J Infect. 2016 May;72(5):544–53 doi:10.1016/j.jinf.2016.02.003 PubMed PMID: 26916042.
- Wasi F, Shuter J. Primary bacterial infection of the myocardium. Front Biosci J Virtual Libr. 2003 May 1;8:s228-231. doi:10.2741/1021 PubMed PMID: 12700039.
- Chang FY, MacDonald BB, Peacock JE, Musher DM, Triplett P, Mylotte JM, et al. A prospective multicenter study of Staphylococcus aureus bacteremia: incidence of endocarditis, risk factors for mortality, and clinical impact of methicillin resistance. Medicine (Baltimore). 2003 Sep;82(5):322–32. doi:10.1097/01.md.0000091185.93122.40 PubMed PMID: 14530781.
- Guo Y, Song G, Sun M, Wang J, Wang Y. Prevalence and Therapies of Antibiotic-Resistance in Staphylococcus aureus. Front Cell Infect Microbiol. 2020;10:107. doi:10.3389/fcimb.2020.00107 PubMed PMID: 32257966; PubMed Central PMCID: PMC7089872.
- Lakhundi S, Zhang K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin Microbiol Rev. 2018 Oct;31(4):e00020-18. doi:10.1128/CMR.00020-18 PubMed PMID: 30209034; PubMed Central PMCID: PMC6148192.
- Adamu Y, Puig-Asensio M, Dabo B, Schweizer ML. Comparative effectiveness of daptomycin versus vancomycin among patients with methicillin-resistant Staphylococcus aureus (MRSA) bloodstream infections: A systematic literature review and meta-analysis. PloS One. 2024;19(2):e0293423. doi:10.1371/journal.pone.0293423 PubMed PMID: 38381737; PubMed Central PMCID: PMC10881006.
- Mlynarczyk-Bonikowska B, Kowalewski C, Krolak-Ulinska A, Marusza W. Molecular Mechanisms of Drug Resistance in Staphylococcus aureus. Int J Mol Sci. 2022 Jul 22;23(15):8088 doi:10.3390/ijms23158088 PubMed PMID: 35897667; PubMed Central PMCID: PMC9332259.
- Kaushik A, Kest H, Sood M, Steussy BW, Thieman C, Gupta S. Biofilm Producing Methicillin-Resistant Staphylococcus aureus (MRSA) Infections in Humans: Clinical Implications and Management. Pathog Basel Switz. 2024 Jan 15;13(1):76. doi:10.3390/pathogens13010076 PubMed PMID: 38251383; PubMed Central PMCID: PMC10819455.
- Pereira JM, Xu S, Leong JM, Sousa S. The Yin and Yang of Pneumolysin During Pneumococcal Infection. Front Immunol. 2022;13:878244. doi:10.3389/fimmu.2022.878244 PubMed PMID: 35529870; PubMed Central PMCID: PMC9074694.
- Anderson R, Feldman C. The Global Burden of Community-Acquired Pneumonia in Adults, Encompassing Invasive Pneumococcal Disease and the Prevalence of Its Associated Cardiovascular Events, with a Focus on Pneumolysin and Macrolide Antibiotics in Pathogenesis and Therapy. Int J Mol Sci. 2023 Jul 3;24(13):11038. doi:10.3390/ijms241311038 PubMed PMID: 37446214; PubMed Central PMCID: PMC10341596.
- Beno SM, Riegler AN, Gilley RP, Brissac T, Wang Y, Kruckow KL, et al. Inhibition of Necroptosis to Prevent Long-term Cardiac Damage During Pneumococcal Pneumonia and Invasive Disease. J Infect Dis. 2020 Nov 9;222(11):1882–93. doi:10.1093/infdis/jiaa295 PubMed PMID: 32492702; PubMed Central PMCID: PMC7896277.
- Dhuriya YK, Sharma D. Necroptosis: a regulated inflammatory mode of cell death. J Neuroinflammation. 2018 Dec;15(1):199. doi:10.1186/s12974-018-1235-0
- Calder PC, Bosco N, Bourdet-Sicard R, Capuron L, Delzenne N, Doré J, et al. Health relevance of the modification of low grade inflammation in ageing (inflammageing) and the role of nutrition. Ageing Res Rev. 2017 Nov;40:95–119. doi:10.1016/j.arr.2017.09.001 PubMed PMID: 28899766.
- Lane JR, Tata M, Briles DE, Orihuela CJ. A Jack of All Trades: The Role of Pneumococcal Surface Protein A in the Pathogenesis of Streptococcus pneumoniae. Front Cell Infect Microbiol. 2022;12:826264. doi:10.3389/fcimb.2022.826264 PubMed PMID: 35186799; PubMed Central PMCID: PMC8847780.
- Bai B, Xu Y, Chen H. Pathogenic roles of neutrophil-derived alarmins (S100A8/A9) in heart failure: From molecular mechanisms to therapeutic insights. Br J Pharmacol. 2023 Mar;180(5):573–88. doi:10.1111/bph.15998 PubMed PMID: 36464854.
- Nishimura N, Fukuda H. Risk of cardiovascular events leading to hospitalisation after Streptococcus pneumoniae infection: a retrospective cohort LIFE Study. BMJ Open. 2022 Nov 4;12(11):e059713. doi:10.1136/bmjopen-2021-059713 PubMed PMID: 36332949; PubMed Central PMCID: PMC9639073.
- Brissac T, Shenoy AT, Patterson LA, Orihuela CJ. Cell Invasion and Pyruvate Oxidase-Derived H2O2 Are Critical for Streptococcus pneumoniae-Mediated Cardiomyocyte Killing. Infect Immun. 2018 Jan;86(1):e00569-17. doi:10.1128/IAI.00569-17 PubMed PMID: 29061707; PubMed Central PMCID: PMC5736805.
- Subramanian K, Neill DR, Malak HA, Spelmink L, Khandaker S, Dalla Libera Marchiori G, et al. Pneumolysin binds to the mannose receptor C type 1 (MRC-1) leading to anti-inflammatory responses and enhanced pneumococcal survival. Nat Microbiol. 2019 Jan;4(1):62–70. doi:10.1038/s41564-018-0280-x PubMed PMID: 30420782; PubMed Central PMCID: PMC6298590.
- Ray D, Yung R. Immune senescence, epigenetics and autoimmunity. Clin Immunol Orlando Fla. 2018 Nov;196:59–63. doi:10.1016/j.clim.2018.04.002 PubMed PMID: 29654845; PubMed Central PMCID: PMC6548177.
- Lin Y, Yang M, Cheng C, Wu J, Yu B, Zhang X. Age-related dysregulation of CXCL9/10 in monocytes is linked to impaired innate immune responses in a mouse model of Staphylococcus aureus osteomyelitis. Cell Mol Life Sci CMLS. 2024 Jul 13;81(1):300. doi:10.1007/s00018-024-05311-2 PubMed PMID: 39001897; PubMed Central PMCID: PMC11335224.
- Gupta P, Hu Z, Kopparapu PK, Deshmukh M, Sághy T, Mohammad M, et al. The impact of TLR2 and aging on the humoral immune response to Staphylococcus aureus bacteremia in mice. Sci Rep. 2023 May 31;13(1):8850. doi:10.1038/s41598-023-35970-3 PubMed PMID: 37258615; PubMed Central PMCID: PMC10232519.
- Brown AO, Mann B, Gao G, Hankins JS, Humann J, Giardina J, et al. Streptococcus pneumoniae translocates into the myocardium and forms unique microlesions that disrupt cardiac function. PLoS Pathog. 2014 Sep;10(9):e1004383. doi:10.1371/journal.ppat.1004383 PubMed PMID: 25232870; PubMed Central PMCID: PMC4169480.
- de Leau MM, Kuipers RS. Cardiovascular complications of Streptococcus pneumoniae bacteraemia. BMJ Case Rep. 2021 Apr 7;14(4):e240341. doi:10.1136/bcr-2020-240341 PubMed PMID: 33827874; PubMed Central PMCID: PMC8030674.
- Reyes LF, Restrepo MI, Hinojosa CA, Soni NJ, Anzueto A, Babu BL, et al. Severe Pneumococcal Pneumonia Causes Acute Cardiac Toxicity and Subsequent Cardiac Remodeling. Am J Respir Crit Care Med. 2017 Sep 1;196(5):609–20. doi:10.1164/rccm.201701-0104OC PubMed PMID: 28614669; PubMed Central PMCID: PMC5620668.
- Dawood H, Nasir S, Khair RM, Dawood M. Infective Endocarditis Secondary to Mycoplasma pneumoniae. Cureus. 2021 Aug;13(8):e17461. doi:10.7759/cureus.17461 PubMed PMID: 34603862; PubMed Central PMCID: PMC8475736.
- Kumar S, Kumar S. Mycoplasma pneumoniae: Among the smallest bacterial pathogens with great clinical significance in children. Indian J Med Microbiol. 2023;46:100480. doi:10.1016/j.ijmmb.2023.100480 PubMed PMID: 37741157.
- Yamamoto T, Okuno M, Kuwano K, Ogura Y. Mycoplasma pneumoniae drives macrophage lipid uptake via GlpD-mediated oxidation, facilitating foam cell formation. Int J Med Microbiol IJMM. 2025 Mar;318:151646. doi:10.1016/j.ijmm.2025.151646 PubMed PMID: 39862618.
- Hu J, Ye Y, Chen X, Xiong L, Xie W, Liu P. Insight into the Pathogenic Mechanism of Mycoplasma pneumoniae. Curr Microbiol. 2022 Dec 2;80(1):14. doi:10.1007/s00284-022-03103-0 PubMed PMID: 36459213; PubMed Central PMCID: PMC9716528.
- Jiang Z, Li S, Zhu C, Zhou R, Leung PHM. Mycoplasma pneumoniae Infections: Pathogenesis and Vaccine Development. Pathog Basel Switz. 2021 Jan 25;10(2):119. doi:10.3390/pathogens10020119 PubMed PMID: 33503845; PubMed Central PMCID: PMC7911756.
- Portugal LR, Fernandes LR, Cesar GC, Santiago HC, Oliveira DR, Silva NM, et al. Infection with Toxoplasma gondii increases atherosclerotic lesion in ApoE-deficient mice. Infect Immun. 2004 Jun;72(6):3571–6. doi:10.1128/IAI.72.6.3571-3576.2004 PubMed PMID: 15155666; PubMed Central PMCID: PMC415665.