Original Article - DOI:10.33594/000000681
Accepted 9 January 2024 - Published online : 22 January 2024
Nitric Oxide Plays a Dual Role in Cardiorenal Syndrome in Vitro Model
bCenter for Natural and Human Sciences (CCNH), Federal University of ABC (UFABC), Santo André, SP, Brazil
Keywords
Abstract
Background/Aims:
Nitric oxide (NO) plays a dual role, acting as both an oxidant and a reducer, with various effects depending on its concentration and environment. Acute kidney injury’s (AKI) pathogenesis observed in cardiorenal syndrome 3 (CRS 3) involves inflammatory responses and the production of reactive oxygen and nitrogen species. However, the role of NO on the development of CRS 3 is still not completely understood. The study aimed to mimic CRS 3 in vitro and investigate NO signaling and inflammatory molecules.Methods:
Thus, HEK293 cells were submitted to normoxia (NX) or hypoxia (HX) protocols for 16 h followed by 3 h of reoxygenation, treated or not with L -NAME. Conditionate medium by HEK293 was transferred to H9c2 for 24 h. Cellular viability was evaluated by MTT assay, real time PCR was used to analyze gene expression and NO content were evaluated in the intra and extracellular medium by amperimetry.Results:
Carbonic anhydrase 9 (CA9) expression increased 2.9-fold after hypoxia. Hypoxia reduced 18 % cell viability in HEK293 that was restored by L -NAME treatment. The sum of nitrite (NO2-) and S-nitrosothiol (S-NO) fractions in HEK293 cells showed a substantial decrease on NO intracellular content (38 %). Both IL-6 and IL-10 decreased in all groups compared to NX cells. Besides TNF-α and Bax/Bcl2 ratio increased in hypoxia (approximately 120-fold and 600-fold, respectively) and L -NAME restored this effect. Regarding H9c2 cells, the S-NO fractions showed a substantial decrease in extracellular content after HX (17%) that was not restored by L -NAME. IL-1β decreases in cardiac cells treated with conditioned medium from HX/L-NAME.Conclusion:
In conclusion this study highlights the complex interplay of NO and inflammatory factors in hypoxia-induced renal and cardiac cell responses, with potential implications for cardiorenal syndrome.Introduction
The crosstalk between kidney and heart is called cardiorenal syndrome and it is defined as a concomitant worsening of cardiac or renal function [1]. Type 3 CRS is defined as acute renocardiac syndrome, occurring when there is acute kidney injury (AKI) and subsequent development of acute cardiac injury [2]. Some data suggest that cardiac damage can be induced by inflammatory mediators, oxidative stress, and stimulation of neuroendocrine systems shortly after acute kidney injury. However, the pathophysiology of this syndrome is quite complex, and various mechanisms have been proposed in addition to the ones mentioned earlier: mitochondrial alterations, electrolyte disturbances, metabolic acidosis, retention of uremic toxins, hyperkalemia, hypocalcemia, ventricular remodeling, and fibrosis [3, 4].
The pathogenesis of AKI is still not fully elucidated due to its complexity, but the inflammatory response is currently accepted as an important pathogenic component, as well as other factors such as endothelial injury, reactive oxygen species (ROS), such as superoxide (O2-), hydrogen peroxide (H2O2), and hydroxyl ions (OH-), production of reactive nitrogen species (RNS), and production of mediators by sublethal damaged tubular cells [5].
It is well known that the mechanisms of free radical generation normally occur in mitochondria, cell membranes, and the cytoplasm. The term ROS encompasses free radical species along with other species that, although they do not have unpaired electrons, are highly reactive due to their instability. It is interesting to note that ROS can combine with other atoms, thus forming other reactive species, such as in the case of nitric oxide (NO), which is a free radical centered on the nitrogen atom and reacts mainly with the superoxide radical (O2-), generating peroxynitrite (OONO-) and other harmful products to the organism [6, 7].
NO can act as an oxidant or a reducer depending on the environment in which it is found Also, NO has a dual role, being cytotoxic or protective (depending on its concentration), a vasodilator, and modulating inflammatory or anti-inflammatory reactions depending on the cell type and stimulus [8]. Its radical (NO-) can be produced in the body by the action of nitric oxide synthases (NOS), from arginine, oxygen, and NADPH, also generating NADP+ and citrulline [9, 10]. However, NO or its reaction products with molecular oxygen and superoxide radicals can modify different macromolecules such as proteins, lipids, and nucleic acids, producing both physiological and pathophysiological effects. NO is involved in processes such as endothelium-dependent vascular relaxation, macrophage-mediated cytotoxicity, inhibition of platelet activation, adhesion, and aggregation, regulation of basal blood pressure and glomerular microcirculation, and it has also been reported to be active in macrophages, neutrophils, kidneys, and myocardium [11].
Considering the aforementioned, it becomes evident that the “conversation” between the heart and the kidney is extensive and permeates numerous cellular, molecular, and physiological processes. Although several studies in the literature have already reported the pathogenesis of type 3 CRS, as well as the role of the inflammatory response in the cardiovascular changes observed in renal injury, systematic study of cellular and molecular mechanisms is essential for the proposition of new clinical interventions and therapeutic approaches [10, 11]. Thus, the aim of the present study was to mimic CRS 3 in vitro and investigate the contribution of NO signaling and inflammatory molecules in renal injury induced by hypoxia.
Materials and Methods
Reagents
Fetal bovine serum (FBS), penicillin/streptomycin, and trypsin were purchased from Gibco (Carlsbad, CA, USA). Dulbecco’s modified Eagle’s medium (DMEM, containing 5.5 mM glucose), methyl-thiazolyl diphenyl tetrazolium bromide (MTT), S-nitrosoglutathione (GSNO), Sulfuric acid (H2SO4), sodium nitrite (NaNO2), potassium iodide (KI), copper chloride (II), phosphate-buffered solution (PBS) and dimethyl sulfoxide (DMSO) were obtained from Sigma-Aldrich (St Louis, MO, USA). Trizol used was from Invitrogen (Waltham, MA, USA) and N(G)-Nitro-L -arginine methyl ester (L -NAME) from Cayman Chemical (Michigan, USA). Ethanol and chloroform were obtained from Synth (Diadema, SP, Brazil). All experiments were carried out using analytical grade water from the Millipore Milli-Q Gradient filtration system and DEPC-treated Water.
Cell Culture and treatments
HEK-293 (Human Embryonic Kidney 293) and H9c2 (Cardiomyoblast) cells were cultivated in high-glucose Dulbecco’s Minimum Essential Medium (DMEM), supplemented with 10 % v/v FBS and 1 % penicillin/streptomycin (100 U/mL). The cells were grown to confluence at 37 °C under 5% CO2 atmosphere and maintained until passages 18. The cells were seeded for experiments at 1 × 105 cells/cm2. Renal cells exposed to NO inhibitor (L -NAME) had the medium immediately changed before hypoxia exposure. L- NAME was added at a non-toxic concentration of 1 mM.
To simulate the renal ischemia/reperfusion protocol, HEK-293 cells were submitted to hypoxia and reoxygenation (H/R). Cardiac and renal cells were cultured until reaching optimal confluence, 80 % for HEK-293 and 60 % for H9c2, respectively, and plated in 6-well plates. HEK-293 cells were exposed to H/R using the BD Gaspak system (BD Diagnostic Systems, USA) for 16 hours and reoxygenation for 3 hours. After placing the culture plate in the BD Gaspak hypoxia system, we also insert a tablet that, as soon as it changes color, indicates that the oxygen inside the bag has reduced to zero. Just after the color change, we start counting the time. Then, the HEK-293 culture medium was transferred to the plate with H9c2 cells and left for 24 hours until collected (Fig. 1).
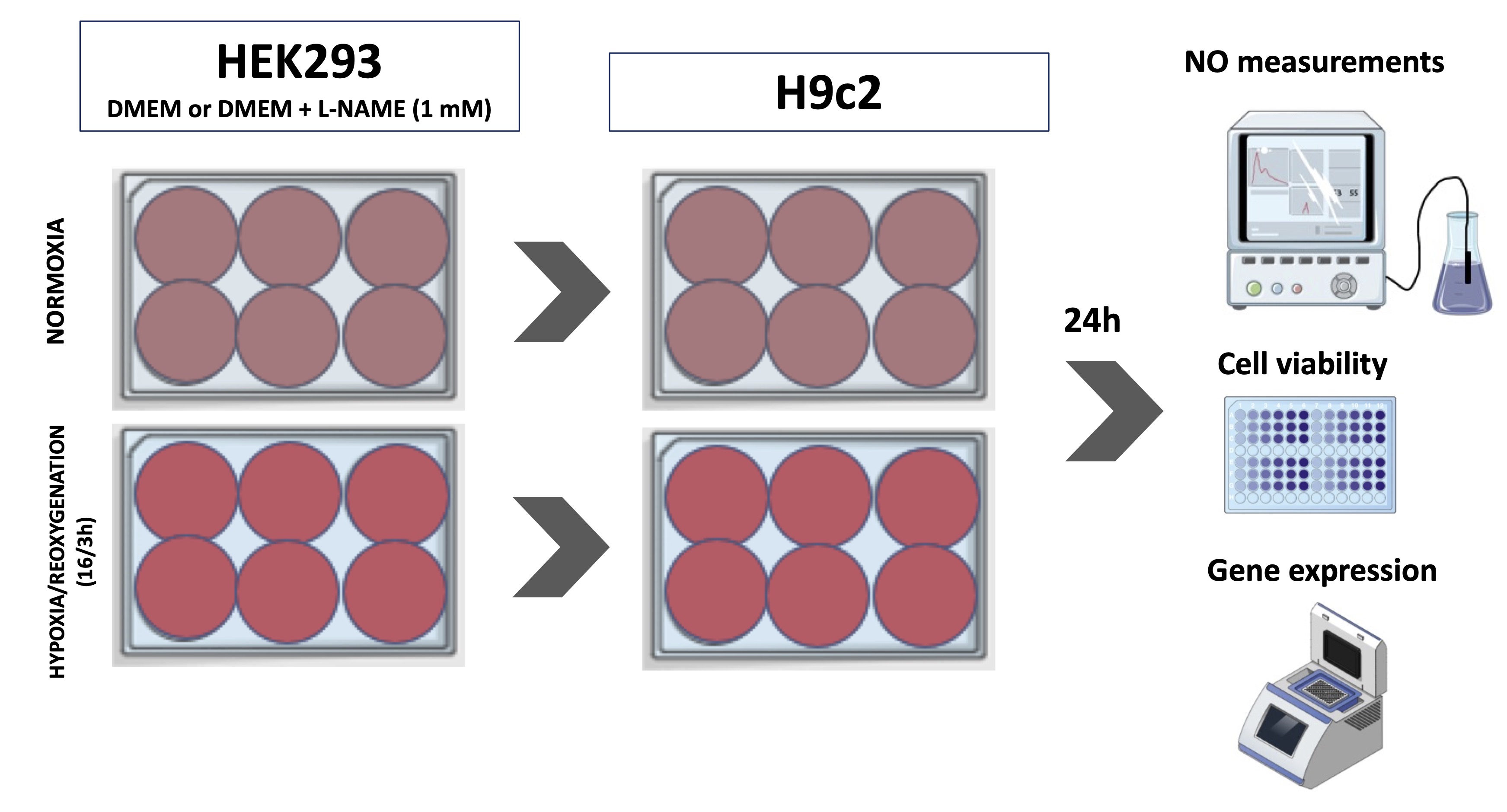
Fig. 1: Experimental design for hypoxia protocol. Cardiac cells (H9c2 - (Cardiomyoblast)) were exposed to medium conditioned by HEK293 (Human Embryonic Kidney 293) cells subjected to hypoxia. L-NAME (N(G)-Nitro-L-arginine methyl ester) treatment was performed to evaluate the role of nitric oxide (NO) on the cellular response. NO measurements, gene expression and cell viability were carried out in both cell types.
Cellular Viability Analysis
For cell viability analysis by MTT, cells were seeded into a 96-well culture plate at a density of 1.0 x 104 cells/well. After H/R for renal cells and after conditioned medium to cardiac cells, the culture medium was removed, and cells were washed 3 times with PBS and incubated with 0.3 mg/ml MTT solution for 2 h at 37°C (in case of HEK293 cells) and 4 h at 37°C (in case of H9c2 cells). After incubation, the medium from each well was carefully removed by aspiration and 200 µL of dimethyl sulfoxide DMSO was added to each well followed by incubation and shaking for 15 s to solubilize the formazan product. Absorbance was measured in a Tecan Infinite M Plex plate reader at 570 nm.
NO measurements
After H/R for HEK-293 cells and 24 h of incubation for H9c2 cells, the cell culture medium was collected (for extracellular NO measurements), the cells were washed and subsequently collected in 500 µL of PBS solution in each well (for intracellular NO measurements). The content was homogenized in the sonicator by short ultrasound pulses (Fisherbrand™ Sound Enclosure for Model 50 and 120 Sonic Dismembrator, USA). Cellular material was collected and stored at -80 °C until use. NOx content was evaluated by quantifying NO2- (Nitrite) and S-NO (S-Nitrosothiol) concentrations through the amperometric technique using the Nitric Oxide TBR 1025 Free Radical Analyzer (World Precision Instruments, Sarasota, FL, USA) as previously reported [12].
Gene expression analysis
For gene expression analysis, total RNA extractions from cells were performed using the TRI Reagent protocol (Sigma-Aldrich, St. Louis, MO, USA). The RNA samples were quantified, and the purities evaluated by the NanoDrop Lite Spectrophotometer (Thermo Scientific, Waltham, USA). The integrity of the RNA (1 µg of total RNA) was evaluated through electrophoresis in a 1% agarose gel, certifying the visualization of the 18S and 28S bands by using UV light in a documentator (Major Science UVDI, Saratoga, USA). Subsequently, 2 ug of RNA were submitted to reverse transcription in a thermocycler (Benchmark’s TC9639 Thermal Cycler, USA) for synthesis of complementary DNA strand (cDNA) using the OligodT primer. Finally, qPCR of samples was performed in duplicate on a real-time thermocycler (Rotor-Gene Q, QIAGEN, USA) and analyzes were performed based on relative gene expression of specific constitutive genes of both cell types using formula 2 -ΔΔCt (where ΔCt = Average Ct of duplicates of the gene of interest in the sample – Average Ct of duplicates of the house keeping gene in the same sample and ΔΔCt = ΔCt - [Average of ΔCt control - ΔCt sample]). For this, the primers were designed using the Primer-BLAST software (NCBI) and synthesized by the company Exxtend (Exxtend, SP). The sequence of all primers used is shown in Table 1.
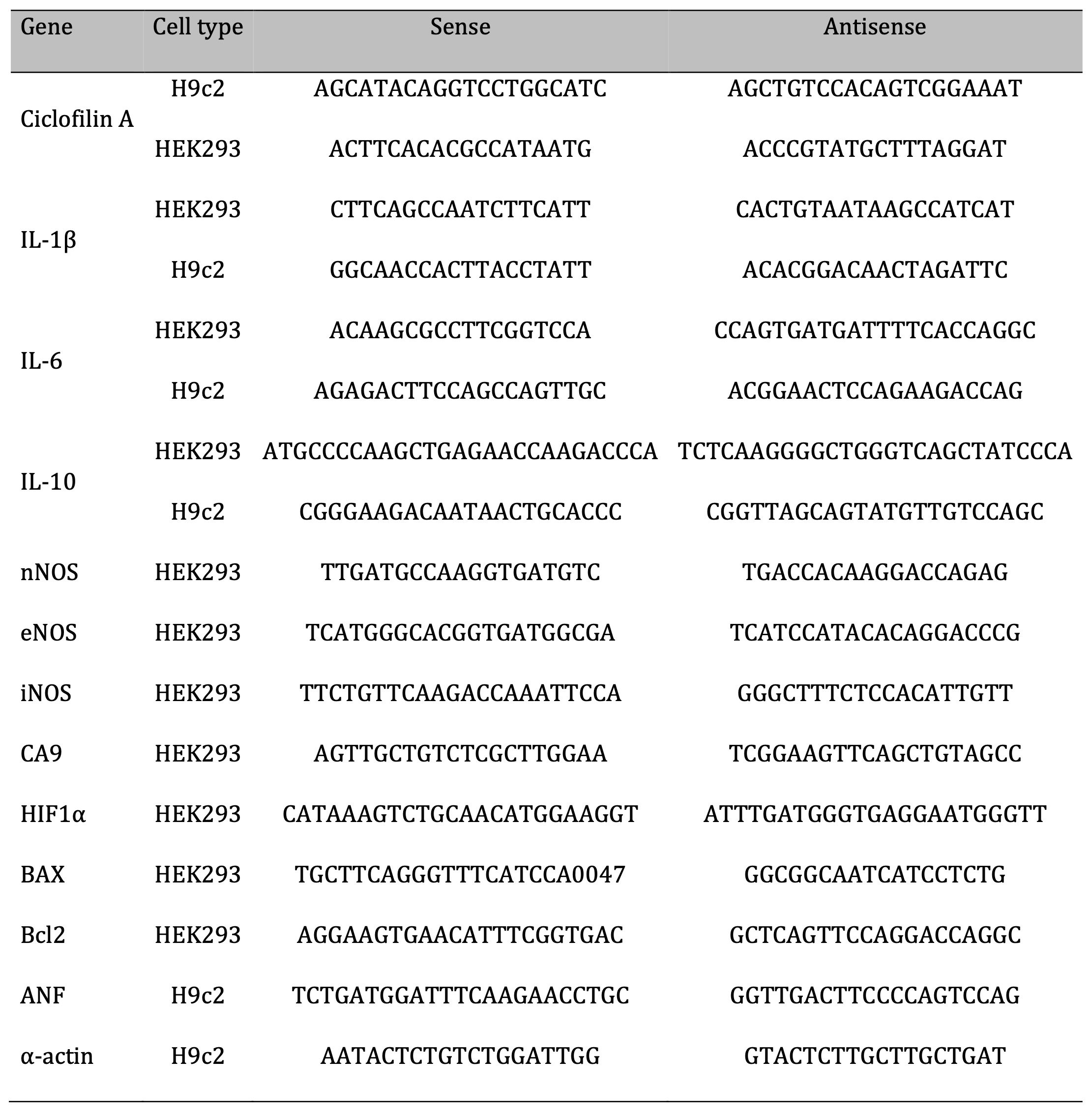
Table 1: List of primers.
Statistical Analysis
All the statistical analysis were performed using GraphPad Prism 8.0 software (San Diego, CA). Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple-comparisons tests or two-way ANOVA followed by Sidak’s post-test. P value < 0.05 was considered significant. All data were shown as mean ± SD of, at least, three replicates.
Results
To assess the occurrence of hypoxia in renal cells, we conducted a gene expression analysis of two hypoxia markers: carbonic anhydrase 9 (CA9) and hypoxia-inducible factor (HIF). As shown in Fig. 2A, there was an increase in CA9 gene expression in the hypoxia-exposed group compared to the normoxia group, indicating signaling and occurrence of hypoxia in renal cells. In parallel, as depicted in Fig. 2B, there was a decrease in HIF-1α gene expression in the hypoxia-exposed groups compared to the normoxia/L -NAME group. Subsequently, to elucidate the potential effects of hypoxia on cellular metabolism and the role of NO in modulating cell viability, we conducted assays to investigate metabolic cytotoxicity in HEK-293 cells. It was possible to observe a significant decrease in cell viability in renal cells subjected to hypoxia (Fig. 2C). Furthermore, cells in normoxia treated with L -NAME exhibited increased cell viability compared to the control group (normoxia). Interestingly, hypoxia-exposed cells treated with L -NAME also showed increased cell viability, as there was no difference between the HX/L-NAME and NX groups, suggesting a cytoprotective effect of L -NAME in both normoxic and hypoxic conditions and a restorative effect in hypoxia condition.
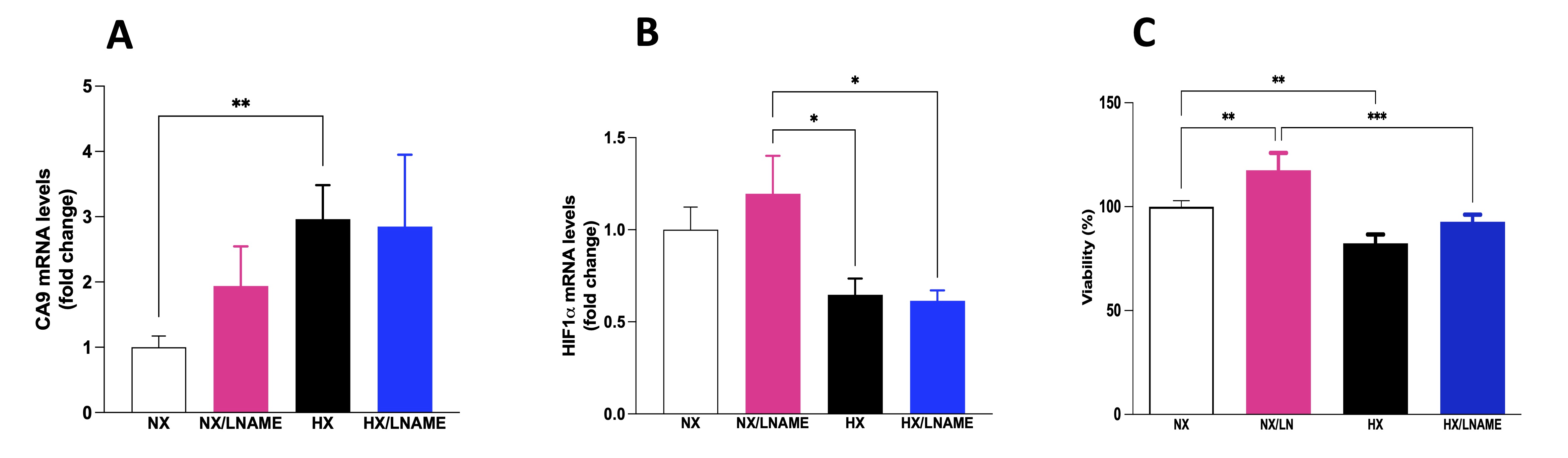
Fig. 2: Effects of hypoxia on HEK293 (Human Embryonic Kidney 293) cells with and without L-NAME treatment. A) CA9 (carbonic anhydrase 9) mRNA levels; B) HIF 1α (hypoxia-inducible factor 1 α ) mRNA levels; C) Cellular viability evaluates by MTT (brometo de 3-[4,5-dimetil-tiazol-2-il]-2,5-difeniltetrazólio) assay. NX: normoxia, NX/L-NAME: normoxia in DMEM + L-NAME, HX: hypoxia, HX/L-NAME: hypoxia in DMEM + L-NAME; n= 3 independent experiments. Data expressed as mean ± standard deviation. * P<0.05; ** P<0.007; *** P<0.0009.
Nitric oxide levels and inflammatory status in HEK-293 submitted to hypoxia
Measurements of nitrite and S-nitrosothiol (S-NO) were performed in both intracellular and extracellular compartments of renal cells subjected to 16 h of hypoxia followed by 3 h of reoxygenation. First, in relation to extracellular nitrite concentration, the results showed that there is no difference between the groups (Fig. 3A). On the other hand, a decrease in extracellular S-NO content was observed in the HX/L -NAME group compared to the NX/L -NAME group (Fig. 3B). Intracellularly, hypoxia reduced nitrite concentration compared to cells exposed to normoxia (Fig. 3D). Additionally, in normoxia, L -NAME inhibited NO synthesis and reduced intracellular nitrite concentration. In contrast, L -NAME treatment in hypoxia restored nitrite levels that had been reduced in normoxia, as there was no difference between the NX and HX/L -NAME groups. However, such reduction was not observed in intracellular S-NO content, which was both reduced during hypoxia and L -NAME treatment conditions (Fig. 3E). Finally, when evaluating the sum of NO2- and S-NO fractions, a substantial amount of extracellular NO was observed (Fig. 3C), along with low intracellular content (Fig. 3F), which aligns with the low levels of all three NOS isoforms in hypoxia-exposed renal cells (Fig. 3G-I). Additionally, L -NAME treatment did not manage to restore NOS levels to control levels. On the other hand, L -NAME treatment under normoxia led to a significant increase in the eNOS isoform (Fig. 3I), alongside high concentrations of NO secreted into the extracellular medium. This indicates that the blockade of NO in normoxia and hypoxia has opposite effects on this isoform. Regarding iNOS, no increase was observed in the hypoxia-exposed and L -NAME-treated groups (Fig. 3H).
NO plays a crucial role in regulating inflammatory cytokines, modulating the production and activity of these molecules, influencing the development and intensity of inflammatory processes. In this sense, the present study showed a decreased pro-inflammatory cytokine IL-6 after L -NAME usage and under hypoxic conditions (Fig. 4B). Additionally, anti-inflammatory cytokine IL-10 was reduced under the same conditions (Fig. 4C). No significant difference in IL-1β was observed among the evaluated groups (Fig. 4A). In contrast to IL6 and IL10 cytokine results, TNF-α increased in hypoxia-exposed renal cells (Fig. 4D).
Next, we evaluated the proteins Bax and Bcl-2, members of a protein family involved in apoptosis regulation, a programmed cell death process crucial in development, homeostasis, and damaged cell removal in the body. We observed a decrease in the anti-apoptotic protein Bcl-2 in hypoxia-exposed cell group compared to renal cells under normoxia (Fig. 4F). No significant differences in the expression of the pro-apoptotic gene Bax were observed across the 4 experimental groups (Fig. 4E). The Bax/Bcl2 ratio was significantly increased in hypoxia-exposed renal cell group compared to the normoxia and HX/L -NAME groups (Fig. 4G). Hypoxia positively regulates the expression of anti-apoptotic genes like Bcl2, dependent on HIF-1.
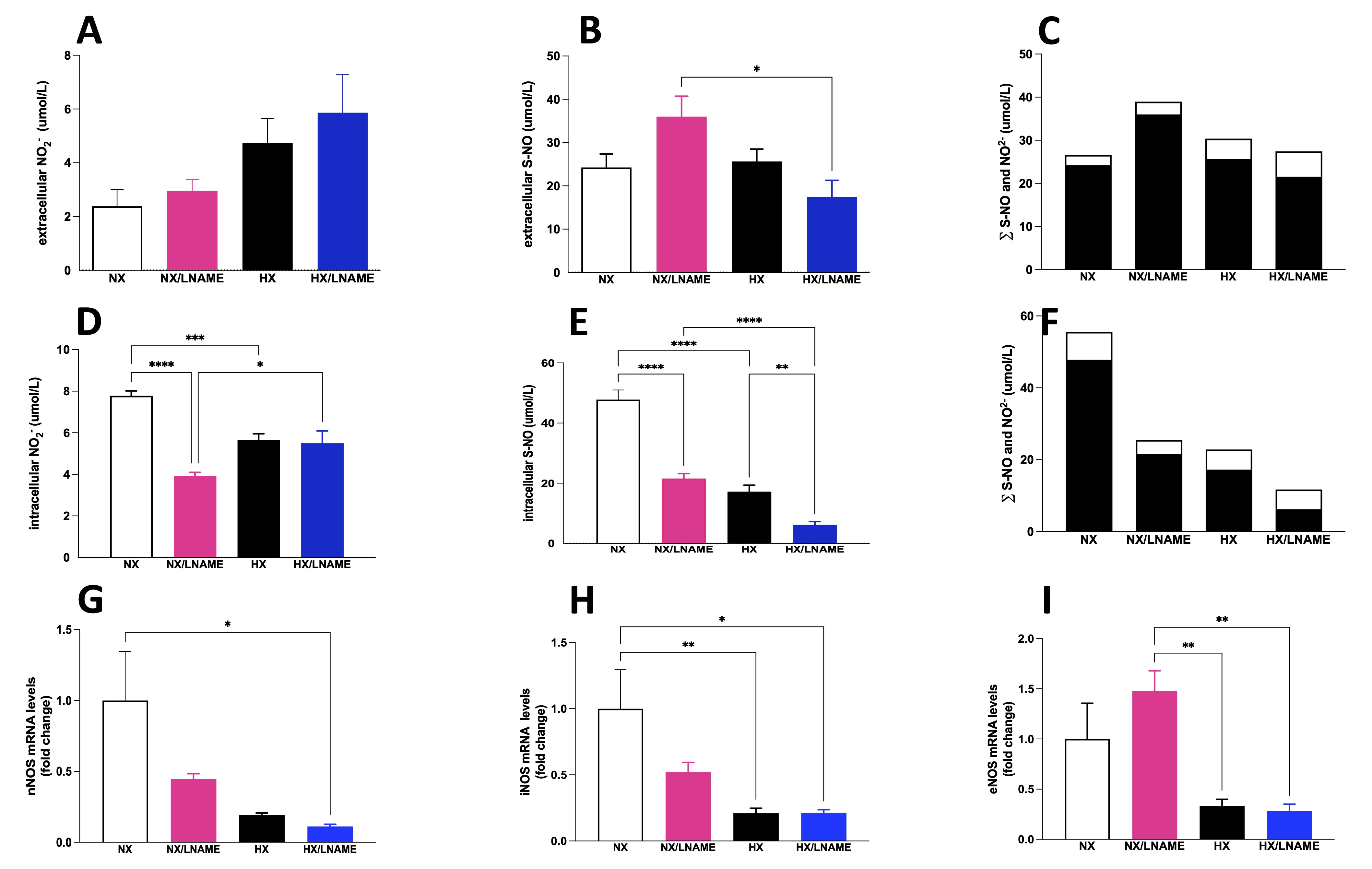
Fig. 3: NO measurement given by NO2- (Nitrite) and S-NO (S-Nitrosothiol) contents in HEK293 (Human Embryonic Kidney 293) cells submitted to hypoxia in the presence or absence of L-NAME treatment. A) Extracellular NO2- (Nitrite) present in culture media; B) Extracellular S-NO (S-Nitrosothiol) present in culture media; C) ∑ of extracellular NO2- (Nitrite) and S-NO (S-Nitrosothiol), and Intracellular (Fig. 3F). The black bars represent the sum of S-NO (S-Nitrosothiol) and white bars, NO2- (Nitrite). In the X axis we showed the groups and Y axis we showed the sum (∑).; D) Intracellular NO2- (Nitrite) present in culture media; B) Intracellular S-NO present in culture media; F) ∑ of intracellular NO2- (Nitrite) and S-NO (S-Nitrosothiol); G) nNOS (Neuronal Nitric Oxide Synthase) mRNA levels; H), iNOS (Inducible Nitric Oxide Synthase) mRNA levels; I) eNOS (Endothelial Nitric Oxide Synthase mRNA levels. NX: normoxia, NX/L-NAME: normoxia in DMEM + L-NAME, HX: hypoxia, HX/L-NAME: hypoxia in DMEM + L-NAME; n= 3 independent experiments. Data expressed as mean ± standard deviation. * P<0.05; ** P<0.007; *** P<0.0009, **** P<0.0001.
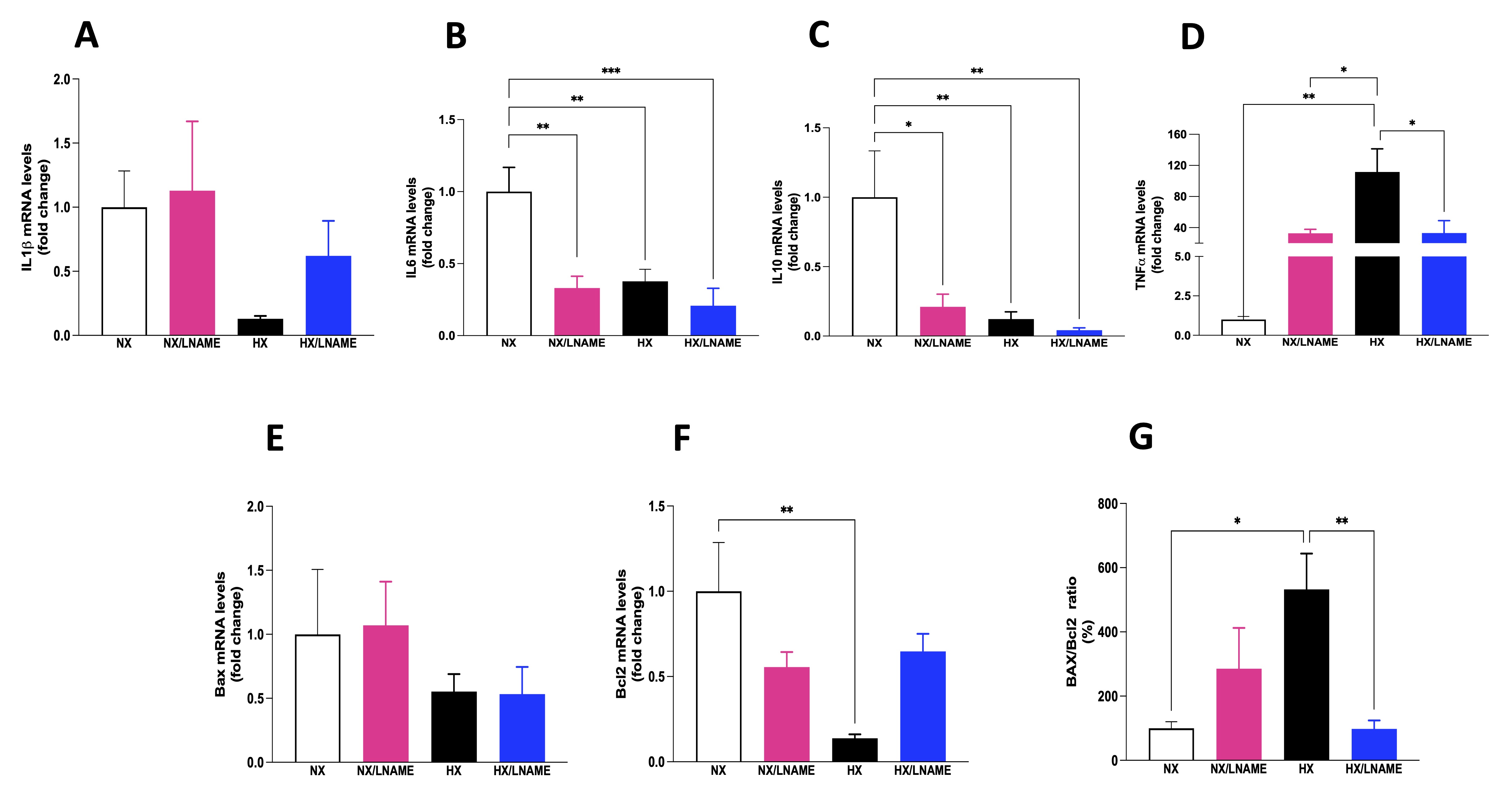
Fig. 4: Inflammatory cytokines status and indicators of cell death in HEK293 (Human Embryonic Kidney 293 cells submitted to hypoxia in the presence or absence of L-NAME treatment. A) IL1β (Interleukin-1 beta) mRNA levels; B) IL6 (Interleukin 6) mRNA levels; C) IL10 (Interleukin 10) mRNA levels; D) TNFα (Tumor necrosis factor alfa) mRNA levels; E) Bax (apoptosis regulator) mRNA levels, F) Bcl2 (B-cell lymphoma 2) mRNA levels; G) Bax/Bcl2 ratio. NX: normoxia, NX/L-NAME: normoxia in DMEM + L-NAME, HX: hypoxia, HX/L-NAME: hypoxia in DMEM + L-NAME; n= 3 independent experiments. Data expressed as mean ± standard deviation. * P<0.05; ** P<0.007.
Effect of HEK-293 - conditionate medium on viability and NO levels of H9c2 cells
To assess the influence of conditioned medium from hypoxia-exposed renal cells on the viability of cardiac cells, culture medium from renal cells was collected and transferred to pre-plated cardiac cells. We conducted viability assays on cardiac cells at two different time points: 30 minutes after supernatant transfer (acute phase) and after 24 h of exposure to the medium (late phase). In the acute phase, we observed decreased viability of cells treated with conditioned medium from hypoxia-exposed renal cells (Fig. 5A). Furthermore, there was no significant difference between normoxia and hypoxia/L -NAME groups, suggesting that L -NAME under hypoxia restored cell viability to control (normoxia) levels, which had decreased under hypoxia. Cardiac cells exposed to conditioned medium for 24 h showed no significant difference in cell viability between normoxia and hypoxia groups (Fig. 5B). Moreover, L -NAME treatment reduced cell viability in H9c2 cells treated with conditioned medium under both normoxia and hypoxia, compared to control cells (normoxia).
Subsequently, we conducted the same analyses for nitrite and S-NO measurements in cellular extracts (intracellular) as well as in the culture medium (extracellular) with cardiac cells conditioned in the supernatant of renal cells after 24 h. Concerning extracellular content, an increase in nitrite concentration was observed in the supernatant of cardiac cells receiving conditioned medium under hypoxia + L -NAME (Fig. 6A). Furthermore, when assessing the sum of NO2- and S-NO fractions, a dual role of L -NAME was evident, antagonistically modulating intracellular NO2- and S-NO levels under hypoxia (Fig. 6D and E). These data indicate a potential balance in NO levels due to the regulation of its NO2- and S-NO subproducts. There was a decrease in total NO content with L -NAME use under normoxia and an increase in total NO content when comparing the HX/L -NAME group to HX (Fig. 6C and F). This pattern may suggest renal cell signaling and NO production stimulation under hypoxia and synthesis blockade.
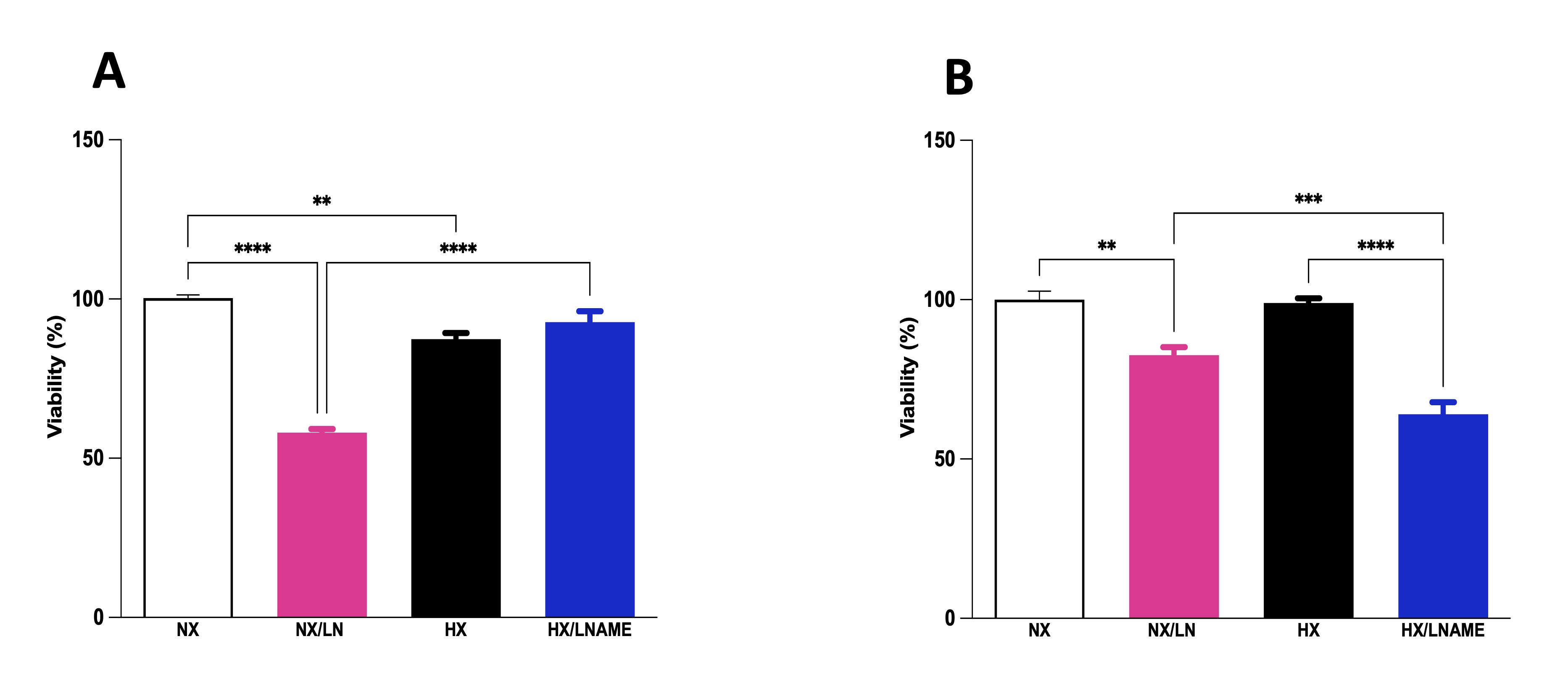
Fig. 5: Effect of conditionate medium by HEK293 (Human Embryonic Kidney 293 on cellular viability of H9c2 (Cardiomyoblast) cells. A) H9c2 cells exposed for 30 minutes (acute phase) to medium conditioned by HEK293, B) H9c2 cells exposed for 24 hours (late phase) to medium conditioned by HEK293. NX: normoxia, NX/L-NAME: normoxia in DMEM + L-NAME, HX: hypoxia, HX/L-NAME: hypoxia in DMEM + L-NAME; n= 4 independent experiments. Data expressed as mean ± standard deviation. ** P<0.007; *** P<0.0009, **** P<0.0001.
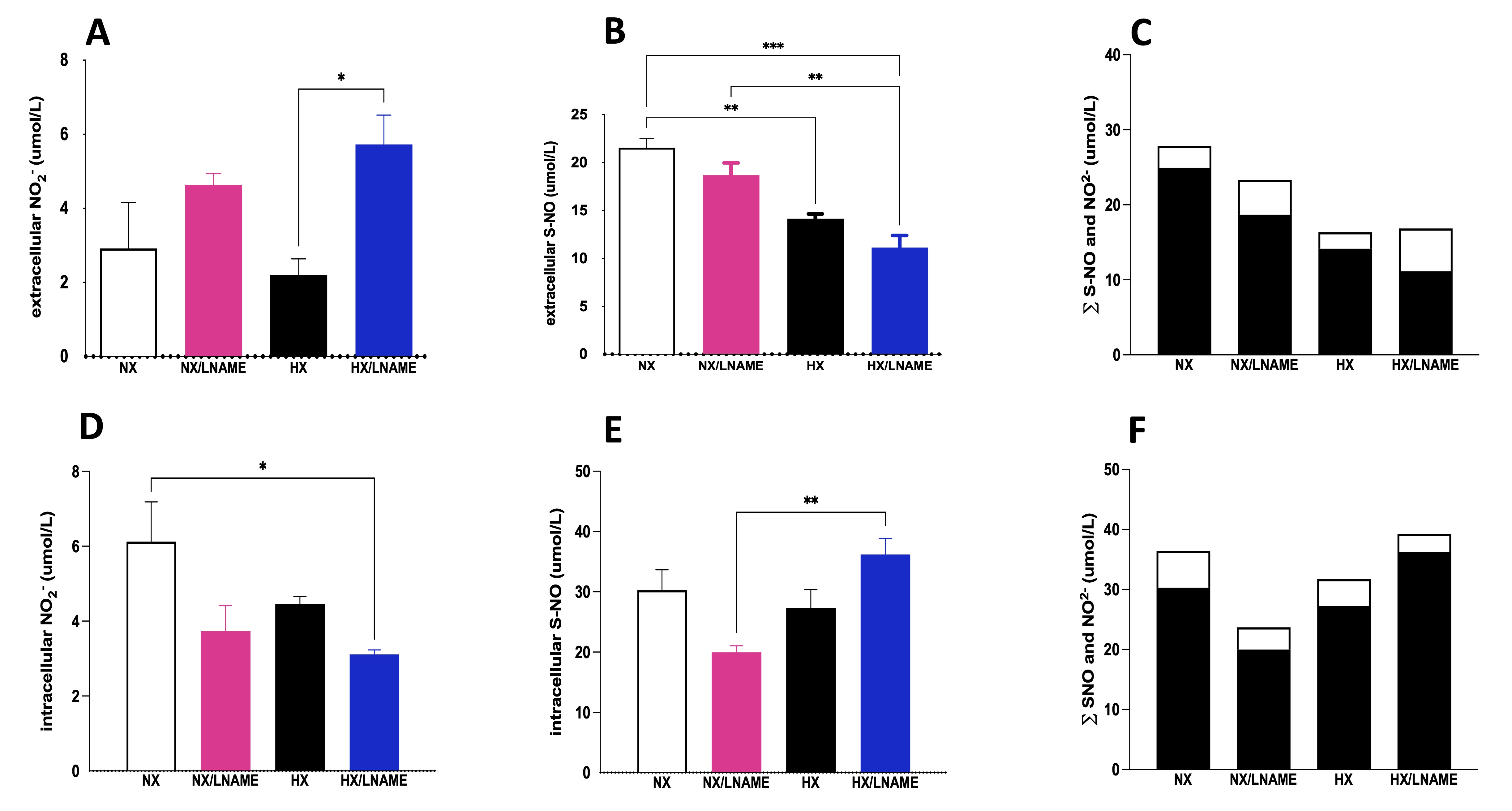
Fig. 6: NO measurement given by NO2- (Nitrite) and S-NO (S-Nitrosothiol contents in H9c2 cells submitted to conditionate medium by HEK293 cells submitted to hypoxia, in the presence or absence of L-NAME treatment. A) Extracellular NO2- (Nitrite) present in culture media; B) Extracellular S-NO (S-Nitrosothiol) present in culture media; C) ∑ of extracellular NO2- (Nitrite) and S-NO (S-Nitrosothiol); D) Intracellular NO2- (Nitrite) present in culture media; B) Intracellular S-NO (S-Nitrosothiol) present in culture media; F) ∑ of intracellular NO2- (Nitrite) and S-NO (S-Nitrosothiol). The black bars represent the sum of S-NO and white bars, NO2- (Nitrite). NX: normoxia, NX/L-NAME: normoxia in DMEM + L-NAME, HX: hypoxia, HX/L-NAME: hypoxia in DMEM + L-NAME; n= 3 independent experiments. Data expressed as mean ± standard deviation. * P<0.05; ** P<0.007; *** P<0.0009, **** P<0.0001.
Modulation of inflammatory status in cardiac cells in hypoxia model-induced cardiorenal syndrome in vitro
In a similar manner as before, the expression of inflammatory cytokines was evaluated in cardiac cells after 24-h treatment with conditioned medium from renal cells. Among the results obtained, there was a decrease in the pro-inflammatory cytokine IL-1β in the group of cardiac cells exposed to conditioned medium from hypoxia-treated renal cells with L -NAME (Fig. 7A). Conversely, there was no significant difference between experimental groups for the pro-inflammatory cytokine IL-6 and the anti-inflammatory cytokine IL-10 (Fig. 7B and C, respectively). Furthermore, modulation of trophic markers in cardiac cells could not be observed in HX group, although L -NAME under hypoxia promoted an increase in α -actin (Fig. 7D). It would be intriguing to evaluate these cells over an extended period to better understand the role of hypoxia-conditioned medium on trophic and cardiac inflammation markers.
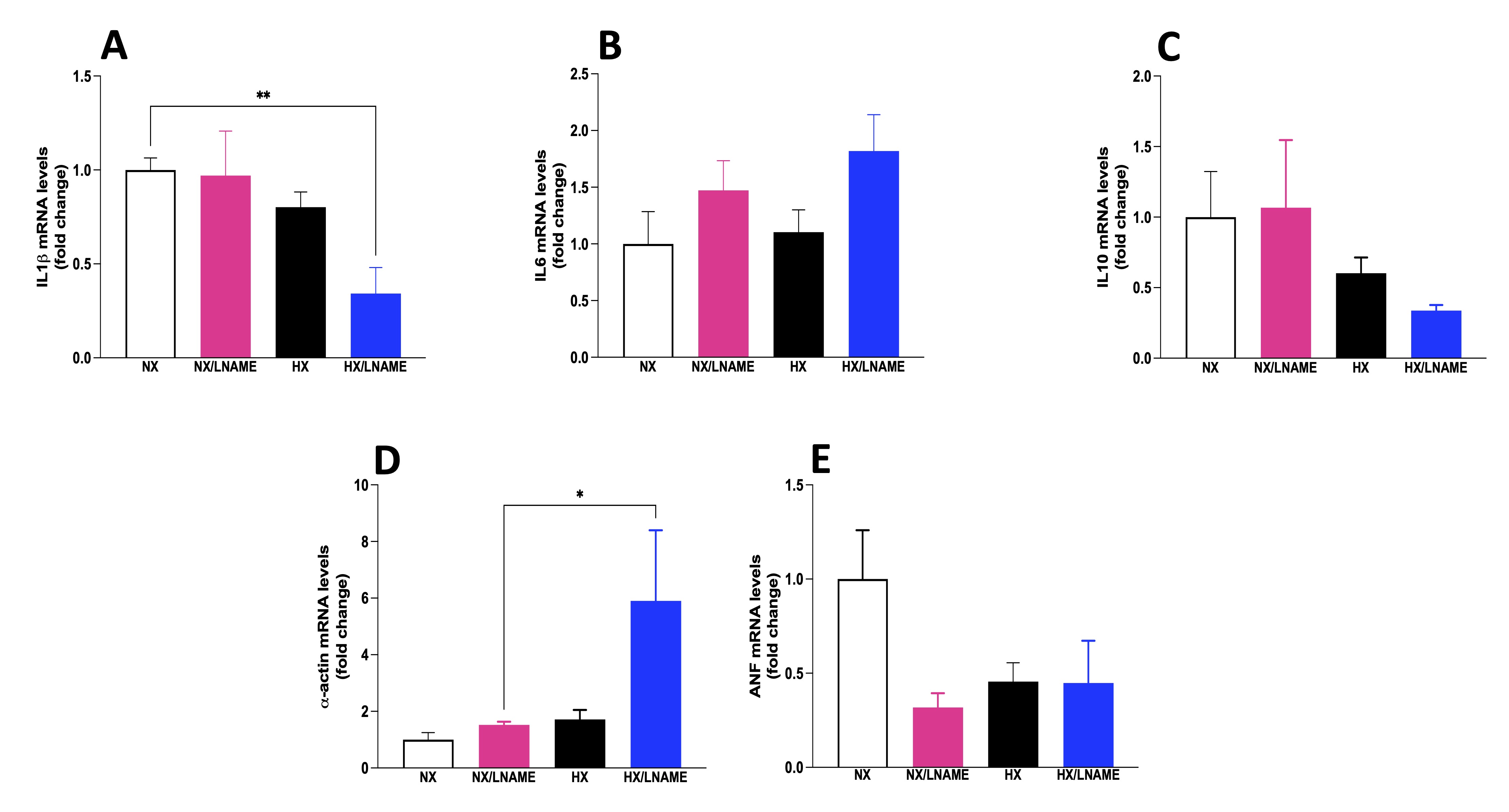
Fig. 7: Inflammatory cytokines status in H9c2 cells cells submitted to conditionate medium by HEK293 in hypoxia and in the presence or absence of L-NAME treatment. A) IL1β (Interleukin-1 beta) mRNA levels; B) IL6 (Interleukin 6) mRNA levels; C) IL10 (Interleukin 10) mRNA levels; D) α-actin mRNA levels; E) ANF (Atrial Natriuretic Factor) mRNA levels. NX: normoxia, NX/L-NAME: normoxia in DMEM + L-NAME, HX: hypoxia, HX/L-NAME: hypoxia in DMEM + L-NAME; n= 3 independent experiments. Data expressed as mean ± standard deviation. * P<0.05.
Discussion
Herein, we showed that NO plays a dual role in CRS in vitro model once we observed a protective response in stress and inflammation situations and stimulating chronic inflammation and cellular damage.
To confirm the success of the experimental protocol, we evaluated two important hypoxia markers. First, hypoxia-inducible factor-1 alpha (HIF-1α) is a heterodimeric transcription factor that senses decreased oxygen availability and responds by transcribing hypoxia-inducible genes involved in angiogenesis, erythropoiesis, energy metabolism, and cellular survival decisions [13]. In many cell types, mRNA levels of HIF-1α and HIF-1β are highly expressed. However, under normoxia, HIF-1α protein is maintained at a low or undetectable level due to continuous proteasomal degradation, while HIF-1β protein is constitutively present [14]. Although HIF-1α has been recognized as a predominant transcriptional activity under hypoxic conditions, it is transiently and rapidly degraded upon increased O2 concentration. Therefore, we hypothesized that the 3-hour reoxygenation period was sufficient to cause proteasomal degradation and generate low levels of gene expression of this marker in HEK293 cells subjected to hypoxia. Second, carbonic anhydrase 9 (CA9) is a transmembrane protein expressed in a wide variety of tumor types and induced by hypoxia [15]. Additionally, CA9 is one of the best-characterized targets of HIF-1, with its promoter containing an essential element for increased transcription during hypoxia [16].
It’s well-known that NO is cardioprotective after hypoxia/reoxygenation-induced injury, through functional and pathophysiological alterations [17]. Endothelial NOS (eNOS) produces physiological NO levels, while inducible NOS (iNOS) produces high NO concentrations [18]. Other studies demonstrated significantly increased eNOS levels immediately after hypoxia, while iNOS activity was induced throughout reoxygenation. This suggests that not only short-term hypoxia but also subsequent reoxygenation positively regulate the NO/NOS system [19]. It is important to note that responses observed in L -NAME-treated cells under hypoxia could be complex and dependent on factors including cellular redox state, NO-related enzyme activity, and protein expression involved in NO signaling pathways. Another study showed cells subjected to hypoxia or glucose followed by reperfusion significantly increased extracellular nitrite production [20]. Additionally, the present study demonstrated NO production induced by 120 minutes of hypoxia and glucose/reperfusion significantly decreased with L -NAME, corroborating observations in renal cells in the present study.
A decrease in extracellular S-NO content was observed in the HX/L-NAME group compared to the NX/L -NAME group. We hypothesize that, in this control (normoxia) situation, the inhibition of NO itself could act as a stimulus for the cell to produce more NO to maintain survival and basal NO content. This is supported by gene expression data indicating increased eNOS isoform expression under normoxia. Conversely, during hypoxia, this control mechanism might become blurred as cells strive to maintain homeostasis to counteract hypoxia-induced injury. Furthermore, a study investigating NOS regulation under hypoxia demonstrated that both hypoxia and thrombin reduced eNOS expression via the Rho/ROCK pathway in endothelial cells [21]. Additionally, L -NAME treatment did not manage to restore NOS levels to control levels. On the other hand, L -NAME treatment under normoxia led to a significant increase in the eNOS isoform, alongside high concentrations of NO secreted into the extracellular medium. This indicates that the blockade of NO in normoxia and hypoxia has opposite effects on this isoform. These data were surprising to us since previous data from our group showed an increase in iNOS expression in mice subjected to an ischemia/reperfusion protocol for 8 days [12]. However, two important points should be highlighted: the difference in experimental protocols, in vivo and in vitro , as well as the reoxygenation time for renal tissue (15 days) and cultured cells (3 hours). Interestingly, a study indicated that eNOS and iNOS are regulated by reoxygenation time after acute hypoxia, where eNOS responds immediately by producing NO, causing vasodilation, while iNOS is linked to the second delayed response, potentially involved in some of hypoxia-induced deleterious consequences [22]. Thus, the more apparent eNOS response and absence of increased iNOS expression could be related to acute kidney injury, over a shorter time frame.
The literature demonstrated that animals simultaneously treated with L -NAME and rosuvastatin in a NO deficiency animal model exhibited increased serum NO and reduced L -NAME-induced inflammatory parameters [23]. Conversely, some studies have shown the use of L -NAME as an inducer of renal injury, as NO deficiency has been associated with the progression from acute kidney injury to chronic kidney disease due to hypertension [24]. Furthermore, other studies showed a protective role of NO during hypoxia, with low concentrations of NO intensifying renal injury [25]. This could be explained by the fact that L -NAME inhibits the classic O2-dependent pathway of NO synthesis, while during hypoxia and acidosis, an alternative NO production pathway is activated. Prior studies in the literature have demonstrated [26] that hypoxia creates an acidic environment, which favors the reduction of nitrite to NO regardless of NOS activity, which might be compromised in this situation [26].
The inflammation caused by hypoxia in AKI model attracts immune cells to the injury site, playing a crucial role in the inflammatory response and immune process regulation. Apart from hypoxia, recent evidence suggests that hypoxia-inducible factor-1 (HIF-1) could accumulate and activate due to growth factors, cytokines, hormones, and NO [27]. Previous studies demonstrated the role of cytokines interleukin IL-1β and tumor necrosis factor-α (TNF-α) in regulating HIF-1α stability [27, 28]. Furthermore, while a ROS-sensitive pathway has been reported (15), details of HIF-1 regulation by TNF-α remain unknown. TNF-α is a pro-tumorigenic cytokine involved in NF-κB activation, acting as an upstream mediator linked to cell activation, differentiation, survival, cytokine production [29]. A recent study reported IL-1β and TNF-α as mediators stimulating HIF-1α DNA-binding activity, implying involvement in gene expression modulation during inflammation. IL-1β increased HIF-1α activity through increased protein abundance, while TNF-α effects were attributed to simultaneous activation of certain proteins within the complex [27, 30]. In the inflammatory milieu, TNF-α transiently activates neutrophils and macrophages, leading to O2− release via NADPH oxidase activation [31]. This oxidative burst releases ROS rapidly and is a crucial part of the defense mechanism against invading microbial pathogens and metastasizing tumor cells. While mainly produced by macrophages, mounting evidence suggests other cell types release TNF-α and other inflammatory mediators, amplifying and enhancing the inflammatory response [32, 33]. TNF-α is cytotoxic to glomerular, mesangial, and epithelial cells, capable of inducing direct renal damage. TNF-α also activates NF-κB transcription, which regulates the expression of genes involved in inflammation, oxidative stress, and endothelial dysfunction [23].
Additionally, we evaluated the proteins Bax and Bcl-2, members of a protein family involved in apoptosis process. Our results showed that hypoxia was able to increase Bax/Bcl-2 ratio corroborating other authors [34]. Furthermore, a study demonstrated that L -NAME during hypoxia prevented events leading to mitochondrial DNA fragmentation and increased Bax/Bcl2 ratio [35]. Moreover, the literature reported that TNF-α can modulate the hypoxia-mediated apoptotic response, affecting the Bax/Bcl2 ratio [36, 37].
Regarding the cardiac cells exposed to conditioned medium for 24 h, the results imply compromised metabolic viability of cardiac cells exposed to conditioned medium from renal cells treated with L -NAME. These findings align with other literature where hypoxia/reoxygenation induced by Na2S2O4, a potent oxygen reducer and eliminator, decreased cell viability concentration-dependently in H9c2 cells [38]. Indeed, NO is an important signaling molecule protecting renal and cardiac cells in hypoxia through functional and pathophysiological alterations [39]. In this regard, studies have demonstrated that cardiomyocytes subjected to oxidative stress by H2O2 treatment for 6 h experienced decreased cell viability. Conversely, cardiomyocytes treated with cryptotanshinone (STC), a plant-derived bioactive, were protected against H2O2-induced cell death. These studies comparatively demonstrate protective effects of compounds like L -NAME in hypoxia-induced renal and cardiac injury [40]. Furthermore, oxidative stress is considered a major contributor to cell death in these cells. L -NAME was capable of exerting a cardioprotective role in cardiac cells exposed to conditioned medium from hypoxia for 30 minutes but not 24 h, corroborating other groups that showed that acetylcholine-induced cardioprotection was also abolished by specific NOS inhibition with L -NAME, suggesting that NO’s role is critical depending on the timing of injury or stimulus [41]
The NO levels in H9c2 cells indicated a potential balance in NO levels due to the regulation of its - NO2- and S-NO subproducts. Once again, in terms of total intracellular content, a 35% reduction in NO levels was observed in cells receiving conditioned medium with L -NAME, while a slight increase was seen in cells receiving conditioned medium under HX+L -NAME. These findings were expected, as controlling NO levels is crucial for various cellular processes. Although modulation occurred in both intracellular and extracellular environments, the final adjustment is delicate and dependent on other factors. Based on the data collected so far, L -NAME’s blockade plays an important role in cellular viability and NO level adjustment under stress conditions like the hypoxia model. The use of L -NAME in summing NO fractions extracellular and intracellular, an interesting pattern emerged. This pattern may suggest renal cell signaling and NO production stimulation under hypoxia and synthesis blockade. In general, these results indicate that collectively, NOS regulate NO quantity during H/R, and NO plays a pivotal role in oxygen sensing at the cellular level, dependent on concentration levels, H/R duration, and physiological conditions. It is also worth noting that cardiac cells were not directly exposed to hypoxia and L -NAME but rather indirectly through the conditioned medium from renal cells directly subjected to these conditions. The heightened total NO content in the group of cells treated with conditioned medium from hypoxia-exposed renal cells and treated with L -NAME explains the low expression of the nNOS isoform in HX and HX/L -NAME groups.
Other studies demonstrated a significant effect of L -NAME on the cardiovascular system, and its treatment can lead to reduced heart rate, cardiac output, and stroke volume [42, 43]. However, the effect observed in the HX/L- NAME group in cardiac cells is indirectly due to L -NAME treatment in renal cells. Additionally, studies have shown that L -NAME rapidly clears from plasma, with a half-life of approximately 19.2 minutes [42, 44]. Hence, we can suggest that the effect of L -NAME on renal cells can signal cardiac cells, which were not directly treated with L -NAME. While elevated levels of peroxynitrite are associated with cytokine induction and subsequent impairments in cardiac function, evidence also suggests that physiologically relevant peroxynitrite concentrations can have cardioprotective effects during myocardial injury in I/R. In this context, NO can also mitigate oxidative stress and provide cardioprotection [45].
Finally, NO inhibition can act as a modulator of cytokines in cellular stress situations. NO is a gaseous molecule produced by cells of the immune system, such as macrophages and endothelial cells, among others. It plays a crucial role in regulating the inflammatory response and communication among immune system cells. In situations of cellular stress, such as tissue injuries, infections, or intense inflammatory processes, NO production can increase. NO can act as an inflammatory mediator, stimulating the production and release of pro-inflammatory cytokines like interleukin-1 (IL-1), interleukin-6 (IL-6), and tumor necrosis factor alpha (TNF-α). However, in some cases, excessive NO production can lead to tissue damage and worsen cellular stress. In such scenarios, selective NO inhibition can be beneficial to modulate the inflammatory response and balance the involved cytokines. By inhibiting NO, it is possible to modulate the production of inflammatory cytokines, reducing excessive pro-inflammatory responses and limiting tissue damage. This strategy can be useful in situations where cellular stress is harmful, such as chronic inflammatory diseases, autoimmune disorders, or pathological processes characterized by uncontrolled inflammatory responses.
Conclusion
In summary, cardiac cells are susceptible to various forms of cellular stress that can trigger detrimental inflammatory processes. In this context, NO plays a dual role. On one hand, NO can be produced as a protective response in stress and inflammation situations, aiding in vasodilation and cardiac function regulation. On the other hand, excessive NO levels can contribute to chronic inflammation and cellular damage. Hence, precise modulation of nitric oxide and the inflammatory response in cardiac cells is essential to ensure homeostasis and cardiovascular health (Fig. 8).
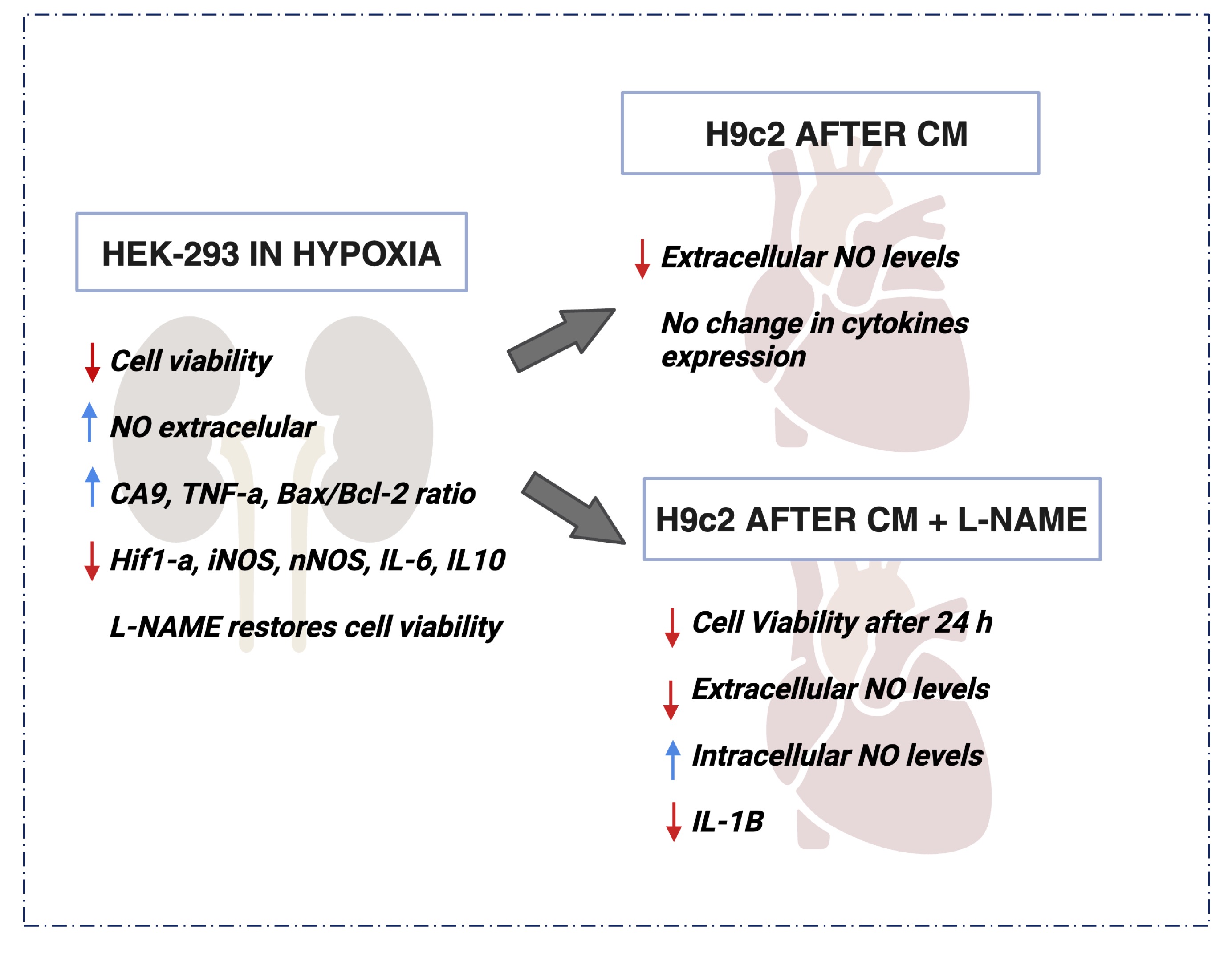
Fig. 8: Outline of the Main Results obtained in this Study. Hypoxia was able to decrease cellular viability and increased the extracellular NO content in HEK293 cells. L-NAME improved the deleterious effect, restoring the viability damaged by hypoxia. On the other hand, H9c2 cells showed a significant decrease on cellular viability and extracellular NO content even after L-NAME treatment. In summary the present study highlights the complex interplay of NO and inflammatory factors in hypoxia-induced renal and cardiac cell responses, with potential implications for cardiorenal syndrome.
Acknowledgements
We have appreciated the financial support from FAPESP (2019/11077-0, 2020/03646-2, 2022/00153-0, 2022/01124-4, 2020/00321-0), CNPq (313117/2019–5 and 406761/2022-1) and CAPES (88887.625462/2021-00). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) – Finance Code 001 (88881.711920/2022-01). Also, we are grateful to the HEK-293 human kidney cells donated by Profa Dra Maria Cristina Carlan da Silva (CCNH, UFABC), and H9c2 mouse heart cells donated by Profa Dra Gabriela Placoná Diniz (ICB, USP).
Disclosure Statement
The authors declare no Disclosure Statement.
References
| 1 | Boudoulas KD, Triposkiadis F, Parissis J, Butler J, Boudoulas H. The Cardio-Renal Interrelationship. Progress in Cardiovascular Diseases. maio de 2017;59:636-648.
https://doi.org/10.1016/j.pcad.2016.12.003 |
| 2 | Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal Syndrome. Journal of the American College of Cardiology. novembro de 2008;52:1527-1539.
https://doi.org/10.1016/j.jacc.2008.07.051 |
| 3 | Neres-Santos RS, Junho CVC, Panico K, Caio-Silva W, Pieretti JC, Tamashiro JA, et al. Mitochondrial Dysfunction in Cardiorenal Syndrome 3: Renocardiac Effect of Vitamin C. Cells. 5 de novembro de 2021;10:3029.
https://doi.org/10.3390/cells10113029 |
| 4 | Bagshaw SM, Hoste EA, Braam B, Briguori C, Kellum JA, McCullough PA, et al. Cardiorenal Syndrome Type 3: Pathophysiologic and Epidemiologic Considerations. Em: McCullough PA, Kellum JA, Mehta RL, Murray PT, Ronco C, organizadores. Contributions to Nephrology Internet. S. Karger AG; 2013 citado 14 de setembro de 2023. p. 137-57. Disponível em: https://www.karger.com/Article/FullText/349971
https://doi.org/10.1159/000349971 |
| 5 | Turgut F, Awad A, Abdel-Rahman E. Acute Kidney Injury: Medical Causes and Pathogenesis. JCM. 3 de janeiro de 2023;12:375.
https://doi.org/10.3390/jcm12010375 |
| 6 | Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol. 1o de março de 1997;82:291-295.
https://doi.org/10.1113/expphysiol.1997.sp004024 |
| 7 | Niki E. Oxidant-specific biomarkers of oxidative stress. Association with atherosclerosis and implication for antioxidant effects. Free Radical Biology and Medicine. maio de 2018;120:425-440.
https://doi.org/10.1016/j.freeradbiomed.2018.04.001 |
| 8 | Modlinger PS, Wilcox CS, Aslam S. Nitric oxide, oxidative stress, and progression of chronic renal failure. Seminars in Nephrology. julho de 2004;24:354-365.
https://doi.org/10.1016/j.semnephrol.2004.04.007 |
| 9 | Shnayder NA, Petrova MM, Popova TE, Davidova TK, Bobrova OP, Trefilova VV, et al. Prospects for the Personalized Multimodal Therapy Approach to Pain Management via Action on NO and NOS. Molecules. 22 de abril de 2021;26:2431.
https://doi.org/10.3390/molecules26092431 |
| 10 | Shnayder NA, Petrova MM, Moskaleva PV, Shesternya PA, Pozhilenkova EA, Nasyrova RF. The Role of Single-Nucleotide Variants of NOS1, NOS2, and NOS3 Genes in the Comorbidity of Arterial Hypertension and Tension-Type Headache. Molecules. 12 de março de 2021;26:1556.
https://doi.org/10.3390/molecules26061556 |
| 11 | Tian Y, Luo J, Xu Q, Liu Y, Cai R, Zhou MS. Macrophage depletion protects against endothelial dysfunction and cardiac remodeling in angiotensin II hypertensive mice. Clinical and Experimental Hypertension. 17 de novembro de 2021;43:699-706.
https://doi.org/10.1080/10641963.2021.1945075 |
| 12 | Caio-Silva W, Da Silva Dias D, Junho CVC, Panico K, Neres-Santos RS, Pelegrino MT, et al. Characterization of the Oxidative Stress in Renal Ischemia/Reperfusion-Induced Cardiorenal Syndrome Type 3. BioMed Research International. 9 de outubro de 2020;2020:1-11.
https://doi.org/10.1155/2020/1605358 |
| 13 | Zhu H, Jackson T, Bunn HF. Detecting and responding to hypoxia. Nephrology Dialysis Transplantation. janeiro de 2002;17:3-7.
https://doi.org/10.1093/ndt/17.suppl_1.3 |
| 14 | Zhou J, Schmid T, Brüne B. Tumor Necrosis Factor-α Causes Accumulation of a Ubiquitinated Form of Hypoxia Inducible Factor-1α through a Nuclear Factor-κB-Dependent Pathway. MBoC. junho de 2003;14:2216-2225.
https://doi.org/10.1091/mbc.e02-09-0598 |
| 15 | Haddad JJ, Land SC. A non‐hypoxic, ROS‐sensitive pathway mediates TNF‐α‐dependent regulation of HIF‐1α. FEBS Letters. 14 de setembro de 2001;505:269-274.
https://doi.org/10.1016/S0014-5793(01)02833-2 |
| 16 | Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 15 de dezembro de 2000;60:7075-7083.
|
| 17 | Chen G, Zhang F, Wang L, Feng Z. Isoflurane alleviates hypoxia/reoxygenation induced myocardial injury by reducing miR-744 mediated SIRT6. Toxicol Mech Methods. maio de 2022;32:235-242.
https://doi.org/10.1080/15376516.2021.1995556 |
| 18 | Wang J, Ji SY, Liu SZ, Jing R, Lou WJ. Cardioprotective effect of breviscapine: inhibition of apoptosis in H9c2 cardiomyocytes via the PI3K/Akt/eNOS pathway following simulated ischemia/reperfusion injury. Pharmazie. setembro de 2015;70:593-597.
|
| 19 | Rus A, Del Moral ML, Molina F, Peinado MA. Upregulation of cardiac NO/NOS system during short-term hypoxia and the subsequent reoxygenation period. Eur J Histochem. 17 de maio de 2011;55:e17.
https://doi.org/10.4081/ejh.2011.e17 |
| 20 | Vahabzadeh G, Soltani H, Barati M, Golab F, Jafari-Sabet M, Safari S, et al. Noscapine protects the H9c2 cardiomyocytes of rats against oxygen-glucose deprivation/reperfusion injury. Mol Biol Rep. agosto de 2020;47:5711-5719.
https://doi.org/10.1007/s11033-020-05549-6 |
| 21 | Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2 de julho de 2002;106:57-62.
https://doi.org/10.1161/01.CIR.0000020682.73694.AB |
| 22 | Rus A, Peinado MÁ, Castro L, Del Moral ML. Lung eNOS and iNOS are Reoxygenation Time-Dependent Upregulated After Acute Hypoxia. Anat Rec. junho de 2010;293:1089-1098.
https://doi.org/10.1002/ar.21141 |
| 23 | Girardi JM, Farias RE, Ferreira AP, Raposo NRB. Rosuvastatin prevents proteinuria and renal inflammation in nitric oxide-deficient rats. Clinics. agosto de 2011;66:1457-1462.
https://doi.org/10.1590/S1807-59322011000800025 |
| 24 | Peeri M, Habibian M, Azarbayjani MA, Hedayati M. Protective Effect of Aerobic Exercise Against L-Name-Induced Kidney Damage in Rats. Archives of Industrial Hygiene and Toxicology. 1o de junho de 2013;64:229-235.
https://doi.org/10.2478/10004-1254-64-2013-2260 |
| 25 | Saleh S, El-Demerdash E. Protective effects of L-arginine against cisplatin-induced renal oxidative stress and toxicity: role of nitric oxide. Basic Clin Pharmacol Toxicol. agosto de 2005;97:91-97.
https://doi.org/10.1111/j.1742-7843.2005.pto_114.x |
| 26 | Pieretti JC, Junho CVC, Carneiro-Ramos MS, Seabra AB. H2S- and NO-releasing gasotransmitter platform: A crosstalk signaling pathway in the treatment of acute kidney injury. Pharmacological Research. novembro de 2020;161:105121.
https://doi.org/10.1016/j.phrs.2020.105121 |
| 27 | El Awad B, Kreft B, Wolber EM, Hellwig-Bürgel T, Metzen E, Fandrey J, et al. Hypoxia and interleukin-1beta stimulate vascular endothelial growth factor production in human proximal tubular cells. Kidney Int. julho de 2000;58(1):43-50.
https://doi.org/10.1046/j.1523-1755.2000.00139.x |
| 30 | Hellwig-Bürgel T, Rutkowski K, Metzen E, Fandrey J, Jelkmann W. Interleukin-1beta and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood. 1o de setembro de 1999;94:1561-1567.
https://doi.org/10.1182/blood.V94.5.1561.417a06_1561_1567 |
| 31 | Klebanoff SJ, Vadas MA, Harlan JM, Sparks LH, Gamble JR, Agosti JM, et al. Stimulation of neutrophils by tumor necrosis factor. J Immunol. 1o de junho de 1986;136:4220-4225.
https://doi.org/10.4049/jimmunol.136.11.4220 |
| 32 | Haddad JJE, Safieh-Garabedian B, Saadé NE, Kanaan SA, Land SC. Chemioxyexcitation (Δp O2/Ros)-Dependent Release Of Il-1β, Il-6 And Tnf-Α: Evidence Of Cytokines As Oxygen-Sensitive Mediators In The Alveolar Epithelium. Cytokine. fevereiro de 2001;13:138-147.
https://doi.org/10.1006/cyto.2000.0789 |
| 33 | Ramseyer VD, Hong NJ, Garvin JL. Tumor necrosis factor α decreases nitric oxide synthase type 3 expression primarily via Rho/Rho kinase in the thick ascending limb. Hypertension. junho de 2012;59:1145-1150.
https://doi.org/10.1161/HYPERTENSIONAHA.111.189761 |
| 34 | Chen JK, Hu LJ, Wang J, Lamborn KR, Kong EL, Deen DF. Hypoxia-Induced BAX Overexpression and Radiation Killing of Hypoxic Glioblastoma Cells. Radiation Research. junho de 2005;163:644-653.
https://doi.org/10.1667/RR3377 |
| 35 | Mishra OP, Randis T, Ashraf QM, Delivoria-Papadopoulos M. Hypoxia-induced Bax and Bcl-2 protein expression, caspase-9 activation, DNA fragmentation, and lipid peroxidation in mitochondria of the cerebral cortex of newborn piglets: The role of nitric oxide. Neuroscience. janeiro de 2006;141:1339-1349.
https://doi.org/10.1016/j.neuroscience.2006.05.005 |
| 36 | Souders CL, Aristizabal-Henao JJ, Patuel SJ, Bowden JA, Zubcevic J, Martyniuk CJ. Interaction between Butyrate and Tumor Necrosis Factor α in Primary Rat Colonocytes. Biomolecules. 30 de janeiro de 2023;13(2):258.
https://doi.org/10.3390/biom13020258 |
| 37 | Shaker FH, El-Derany MO, Wahdan SA, El-Demerdash E, El-Mesallamy HO. Berberine ameliorates doxorubicin-induced cognitive impairment (chemobrain) in rats. Life Sci. 15 de março de 2021;269:119078.
https://doi.org/10.1016/j.lfs.2021.119078 |
| 38 | Lin S, Lin J mao, Zhang L, Chen D xin, Xiao F, Chen H wei, et al. Shexiang Tongxin Dropping Pill (麝香通心滴丸) Protects against Na2S2O4-Induced Hypoxia-Reoxygenation Injury in H9c2 Cells. Chin J Integr Med. junho de 2019;25:439-445.
https://doi.org/10.1007/s11655-018-2976-4 |
| 39 | Chu D, Zhang Z. Trichosanthis Pericarpium Aqueous Extract Protects H9c2 Cardiomyocytes from Hypoxia/Reoxygenation Injury by Regulating PI3K/Akt/NO Pathway. Molecules. 20 de setembro de 2018;23:2409.
https://doi.org/10.3390/molecules23102409 |
| 40 | Shi G, Wang Y, Yang J, Liu T, Luo F, Jin G, et al. Effect of Cryptotanshinone on Measures of Rat Cardiomyocyte Oxidative Stress and Gene Activation Associated with Apoptosis. Cardiorenal Med. 2021;11:18-26.
https://doi.org/10.1159/000507184 |
| 41 | Liu H, McPherson BC, Zhu X, Da Costa MLA, Jeevanandam V, Yao Z. Role of nitric oxide and protein kinase C in ACh-induced cardioprotection. American Journal of Physiology-Heart and Circulatory Physiology. 1o de julho de 2001;281:191-197.
https://doi.org/10.1152/ajpheart.2001.281.1.H191 |
| 42 | Duggan JA, Tabrizchi R. Effect of nitric oxide synthase inhibitor N(omega) nitro-L-arginine methyl ester on relaxant responses to calcium channel antagonists in isolated aortic rings from Dahl normotensive and hypertensive rats. J Cardiovasc Pharmacol. março de 2002;39:354-362.
https://doi.org/10.1097/00005344-200203000-00006 |
| 43 | Bil-Lula I, Krzywonos-Zawadzka A, Sawicka J, Bialy D, Wawrzynska M, Wozniak M, et al. L-NAME improves doxycycline and ML-7 cardioprotection from oxidative stress. Front Biosci (Landmark Ed). 1o de janeiro de 2018;23(2):298-309.
https://doi.org/10.2741/4592 |
| 44 | Avontuur JA, Buijk SL, Bruining HA. Distribution and metabolism of N(G)-nitro-L-arginine methyl ester in patients with septic shock. Eur J Clin Pharmacol. outubro de 1998;54:627-631.
https://doi.org/10.1007/s002280050525 |
| 45 | Dhalla N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovascular Research. 18 de agosto de 2000;47:446-56.
https://doi.org/10.1016/S0008-6363(00)00078-X |