A Model of Interaction Between Apocynin and NADPH Oxidase Enzyme to Analyze the Possible Targets Responsible for Inhibition by Computational Analysis
bDepartment of Biotechnology, School of Engineering and Technology, Sharda University, Greater Noida, Uttar Pradesh, India,
cDepartment of Pharmacognosy, Era College of Pharmacy, Era University, Lucknow, Uttar Pradesh, India,
dDepartment of Pharmacognosy, Amity Institute of Pharmacy, Lucknow, Amity University, Uttar Pradesh, Sector 125, Noida, India
Keywords
Abstract
Background/Aims:
A multi-component enzyme system called NADPH oxidase (NOX) helps innate immunity by generating reactive oxygen species (ROS). NOX hyperactivation has been associated w several diseases. This enzyme is a membrane-bound complex made up of six subunits when it is active. These enzymatic subunits are considered to be potent inhibitors of enzyme activity and good targets for reducing oxidative stress.Methods:
The present study aimed to analyze the possible targets: the different subunits of NOX, for their interactions with apocynin to identify its possible mechanism of inhibition for NOX, using in silico tools. Monomer, dimer, and trimer of apocynin were docked to various subunits of NOX.Results:
Comparable glide scores were obtained when the monomer and dimer of apocynin were docked with p47phox complete subunit of NOX and were better than in comparison to trimer. Free Energy of Binding (FEB) was highest in the case of the trimer (-37.4 Kcal/mol), followed by the dimer (-21.2 Kcal/mol) and monomer (-18.2 Kcal/mol). Dimer obtained the highest glide score of 8.25 (FEB =-25.1 Kcal/mol) with p67phox-isoform 2. The PH domain of p47phox and the SH3 domain of p67phox have their own best binding energy with dimmer. While molecular docking with Rac-Zn-GD, P, dimer, and trimer have shown comparable FEB. The residues, on which the ligands were found to interact, were of major significance being present in those domains that vicinity to inhibit or activate the complex and are important for the protein structure and functioning. MDS studies have confirmed the findings that the Apocynin trimer molecule has superior stability and interactions with the enzyme complex.Conclusion:
It can be concluded from the study that trimer and dimer have better interactions in terms of FEB with p67phox and p47phox, indicating the reported findings in the literature.Introduction
NOX is a multi-component enzyme system that supports innate immunity by producing ROS during phagocytosis [1]. Chronic Granulomatous Disease (CGD), atherosclerosis, pancreatic fibrosis, and some other inflammatory disorders have been linked to NOX hyperactivation [1-4]. In its active form, this enzyme is found as a membrane-bound complex featuring six subunits (Membrane components- Nox2/gp91phox, p22phox, cytosolic components- p47phox, p67phox, p40phox, GTP-Rac) [1, 5]. These enzymatic subunits are thought to be effective targets for inhibiting enzyme function and mitigating oxidative stress or inflammation [2]. Earlier it has been screened and identified as various natural and synthetic inhibitory compounds for NOX [6-9].
In resting cells, p47phox, p67phox, and p40phox establish a ternary complex in the cytoplasm, whereas Rac is complexed with Rho-GDP, the dissociation inhibitor [10]. The synthesis of the catalytic oxidase complex at the membrane is essential for gp91phox /Nox2 activation. Activation involves stimulus-induced membrane translocation of p47phox, p67phox, p40phox, and Rac [11]. The presence of p22phox on stimulation drives the ternary complex to translocate to the membranous system [12]. The PRR domain of the NOX subunit p22phox is largely made up of a proline-rich helix that is responsible for binding to the two SH3 domains of p47phox. The PX domain at the N-terminal end with two neighboring SH3 domains, one AIR domain, and one PRR domain at the C-terminal end make up p47phox, which serves as an integrator for all the subunits [13]. The PX domain and SH3 domains are responsible for translocation to membrane components, whereas the AIR domain is involved in p47phox activation, and the PRR domain, in the end, is responsible for binding p67phox via its lone SH3 domain [10]. It also comprises a TPR domain (also known as an activator) that binds to Rac, an activation domain (AD) that activates NOX, and a PB1 domain sandwiched between two SH3 domains [14]. The P67phox PB1 domain has been reported to be responsible for binding to the p40phox PB1 domain [10] which suggests that p40phox may aid in the recruitment of p47phox and p67phox to membranes. It also has a PX domain that binds phosphatidylinositol 3-phosphate as well as a PB1 domain that recruits p67phox. When all the subunits are coupled together, signal transduction occurs, and oxygen radicals are generated catalytically at gp91phox /Nox2. These radicals then react with oxygen to yield ROS such as oxygen ions and peroxides such as H2O2. Six more Nox2 homologs have similar functions but are structured differently [5]. P67phox homologs encompass isoforms 1, 2, and 3 as well as p67-like proteins [15]. As previously noted, NOX has been inhibited by a variety of natural and synthetic compounds. To design/screen potential inhibitors for NOX, all subunits can presumably be targeted. Apocynin has been around for a long time, and its clinical utility has been studied both in vivo (in rats) and in vitro [16]. Interestingly, apocynin inhibits the NOX activity meanwhile; it does not obstruct the phagocytic or any other defense roles of granulocytes [17]. This was discovered in Canadian hemp, and it was later discovered in Picrorhiza kurroa, which is now a major source of this compound All subunits can presumably be targeted to design/screen potential inhibitors for NOX [16]. It has been demonstrated to be an efficient antioxidant, anti-inflammatory, hepatoprotective, anti-asthmatic, and anti-arthritic activity [4, 17]. When myeloperoxidase (MPO) dimerizes or trimerizes apocynin in the presence of H2O2 [17], it inhibits NOX [17]. Its further functions in MPO-secreting cells, not in MPO-deficient ones. Moreover, it was discovered that if neighboring cells provide MPO, apocynin can still be effective in inhibiting NOX in non-MPnon-MPO-producing [17]. The principal mechanism of apocynin’s inhibitory activity on NOX is thought to be its interference with p47phox and/or p67phox translocation [4, 10, 16, 17]. These investigations do not say if apocynin interacts directly with these subunits or works in the background to prevent the enzyme complex from accumulating on the plasma membrane. Race protein, the sixth component of NOX, has been shown to limit cell migration, which is an essential task of NOX. Such response has been linked to a combination of apocynin oxidation products at pH 8, which could be mostly attributable to the trimer of apocynin [16]. There are possibilities that, they block different sites at different subunits which are ultimately involved in blocking the assembly of the enzymes, therefore it is necessary to analyze the interaction of various Apocynin forms with the various subunits and with the entire family of NOX (isoforms present at different locations in a biological system) to postulate the inhibitory effects in total and thereby finding out their effectiveness in various locations in the body, which will, in turn, help us to deduce the mechanism of apocynin’s action/inhibition [16].
The present study is based on viewing the interaction(s) of different subunits of NOX and related proteins with apocynin. In this study interaction of apocynin with the enzyme subunits was investigated by computational methods. Interactions were seen with the monomer, dimer, and trimer of apocynin (shown in figure 1) with different enzyme subunits separately and in combinations.
Fig. 1: Structure of different forms of Apocynin made using ChemSketch 12.1.
Materials and Methods
In silico Molecular docking
Different forms of Apocynin viz. monomer, dimer, and trimer were prepared using ChemSketch 12.1 [18] and the builder module in Schrödinger [19]. They were then used for blind docking over the sub-units and biologically active domains of NOX enzyme to find the possible interaction sites on the enzyme and then to correlate with their biological functions [20, 21]. Moreover, the different Apocynin forms also satisfied Lipinski’s rule of five. This property shows the drug-likeness of these forms [22]. Molecular Docking of Apocynin forms was carried out on the following proteins/peptides/subunits:
- PX domain of p47phox (PDB ID-1GD5) [23],
- C-terminal SH3 domain of p67phox (From PDB ID-1K4U) [24],
- Complex of the SH3 domain of p67phox with C-terminal tail region of p47phox (PDB ID-1K4U),
- RAC-Zn-GTP complex (PDB ID-2P2L),
- Complete structures of p47phox (predicted from I-TASSER)
- Complete sequence structures p67phox (I-TASSER outputs for: NCF2/ p67phox -ISF1 and NCF2/ p67phox –ISF2, NCF2/ p67phox –ISF3)
Protein Preparation. The PDB files of the required proteins and various complexes (like the complex of p47phox with p67phox) were downloaded from the RCSB protein database (PDB, NCBI) [25]. Complete structures for p47phox, p67phox, and other homologs or isoforms were not available on PDB (Protein database) and they were predicted using the I-TASSER server [26]. In the case of downloaded PDB files, proteins with ligand-free, are cleaned and optimized using the protein preparation wizard tab in Schrödinger [27]. Complete structure outputs of I-Tasser were subjected to optimization and secondary structure prediction using prime tab.
Receptor Grid generation. Receptor Grids were prepared subsequently after concluding downloading of protein structure for blind or site-specific docking. The grids were generated using a Receptor Grid Generation Panel under Glide Tab for the complete protein stretch to find out all probable active sites for all forms of apocynin. The grid size was modified depending on the size of the protein [28].
Ligand Preparation. Ligand structures were prepared using the LigPrep Module (shipped by Schrödinger 2021-2). 2D structures of the Ligands were made using Chemsketch v12.0 and were used as an input for LigPrep. Ionization states were generated at a specific pH range using the ionizer program for use by Glide. Unwanted water molecules and counterions were removed using the Desalt Program of LigPrep [29].
Active Site Molecular Docking
Ligand docking was performed using the Extra Precision (XP) algorithm of Glide (Glide v91117, Schrödinger, LLC, New York, NY, 2021-2) [21, 30, 31]. After it was ensured that, both the protein and the ligands are in suitable forms required for docking, calculations were performed using the Ligand Docking Tab of Glide [21]. Both the receptors and ligands were given as input along with the grid generated. Glide searches for favorable interactions between the receptor and ligand. Glide poses were internally generated and were passed through a series of hierarchical filters that evaluate the interaction of ligands with the receptor [21]. At this stage, the initial filters which use a grid-based method, test the spatial fit of the Ligand to the active site and these filters also examine the complementarity in interactions between the ligands and the receptor. Poses that made it through these initial screens enter the final stage of the algorithm, which includes evaluation and minimization of a grid approximation to the OPLS-AA non-bonded ligand-receptor interaction energy. Energy minimized poses are further exposed to the final scoring. By default, Schrödinger’s GlideScore scoring function is used to score the poses and to generate a single best pose as an output for each input ligand [29, 31].
Gscore = 0.065* vdw + 0.130* Coul + Lipo + Hbond + Metal +BuryP + RotB + Site
ʋ; vdW = van der Waal energy; Coul = Coulomb energy; Lipo = lipophilic contact term; HBond = hydrogen bonding term; Metal=metal-binding term; BuryP = penalty for buried polar groups; RotB = penalty for freezing rotatable bonds; Site = polar interactions at the active site.
Prime MM GBSA and Free Energy of Binding (FEB)
For the calculation of FEB, the MMGBSA panel of Prime is used. Prepared protein and Ligand were used as an input for the Prime MM GBSA program [21, 30]. Output is obtained in the form of a Maestro Structure file and consists of the following properties [32] including coulombic energy, the covalent binding energy of the complex, van der Waals energy of the complex, generalized Born electrostatic solvation energy of the complex, MMGBSA energy of the complex, free Ligand & non-complexed receptor along with MMGBSA energy of receptor-ligand complex [33]. The energy difference is calculated for each of the Ligands and the Receptor using the equation:
ΔE = Prime MMGBSA Complex Energy- Prime MMGBSA Ligand Energy- Prime MMGBSA Receptor Energy
Where, ʋ; ∆E is Free Energy of Binding.
The protein targets were first chosen selectively and were then prepared to be docked with 3 forms of apocynin viz. Monomer, Dimer, Trimer (at two different pH7, pH8) [1].
Molecular Dynamic Simulation
GROMACS-2018.1 has been used to perform Molecular dynamics simulation on the coordinates of protein-ligand complexes [34], and the protein interactions were given with the CHARMM36 force field [35]. CGenFF server has been used to generate the ligand parameters [36]. Each protein-ligand complex has been then solvated with water molecules TIP3P and localized in the center of a cubic simulation box with 10 Å from the edges. The ions (Na+ Cl−), 0.15 M have been added in order to neutralize the simulated system. The boundary condition was specified in all the coordinates i.e. x, y, and z directions [37], the electrostatic interactions were evaluated using particle-Ewald summation [37], and the vdW interactions were calculated using a cut-off of 10 Å. The energy consumption of the generated systems was minimized using the steepest descent and conjugate gradient techniques. For 50, 000 steps, energy minimization was conducted. Equilibration was primarily conducted in an NVT ensemble for 500 ps, and subsequently in an NPT ensemble for another 500 ps. Parrinello–Danadio–Bussi thermostat [38] and a Parrinello–Rahman pressure [39] has been used to set at Temperature of 300 K and pressure of 1 bar, respectively. The integration step was set to 2 fs. The system has been simulated for 100 ns and taking snapshots in every 10 ps for subsequent analysis of the MDS [40, 41].
Results
Docking with the complete structure of p47phox
Optimized protein/subunits and ligands were docked using ligand docking under Glide. Glide generates glide scores, E-model, and other energies like Vdw forces, good contacts, and bad contacts. H-bonds with receptors were viewed using H-bonds to the receptor tab in the Pose viewer submenu. Free energy of binding has been calculated using the Prime-MM GBSA module of Schrödinger which is based on molecular mechanics generalized Born surface area. Figure (2a, 2b) shows the docking results of the Monomer of Apocynin with p47phox complete protein structure. As, it is indicated that, monomer of the apocynin binds on PX domain (Glide score -3.818, E model =-32.3, Prime energy =-15046.47 Kcal/mol, FEB -29.56 Kcal/mol). Compared to monomer, the dimer has a slightly high glide score (Glide score= -4.603, E model= -54.4, Prime energy= 15045.24 Kcal/mol, FEB= -32.67 Kcal/mol) as shown in Figure (2c, 2d).
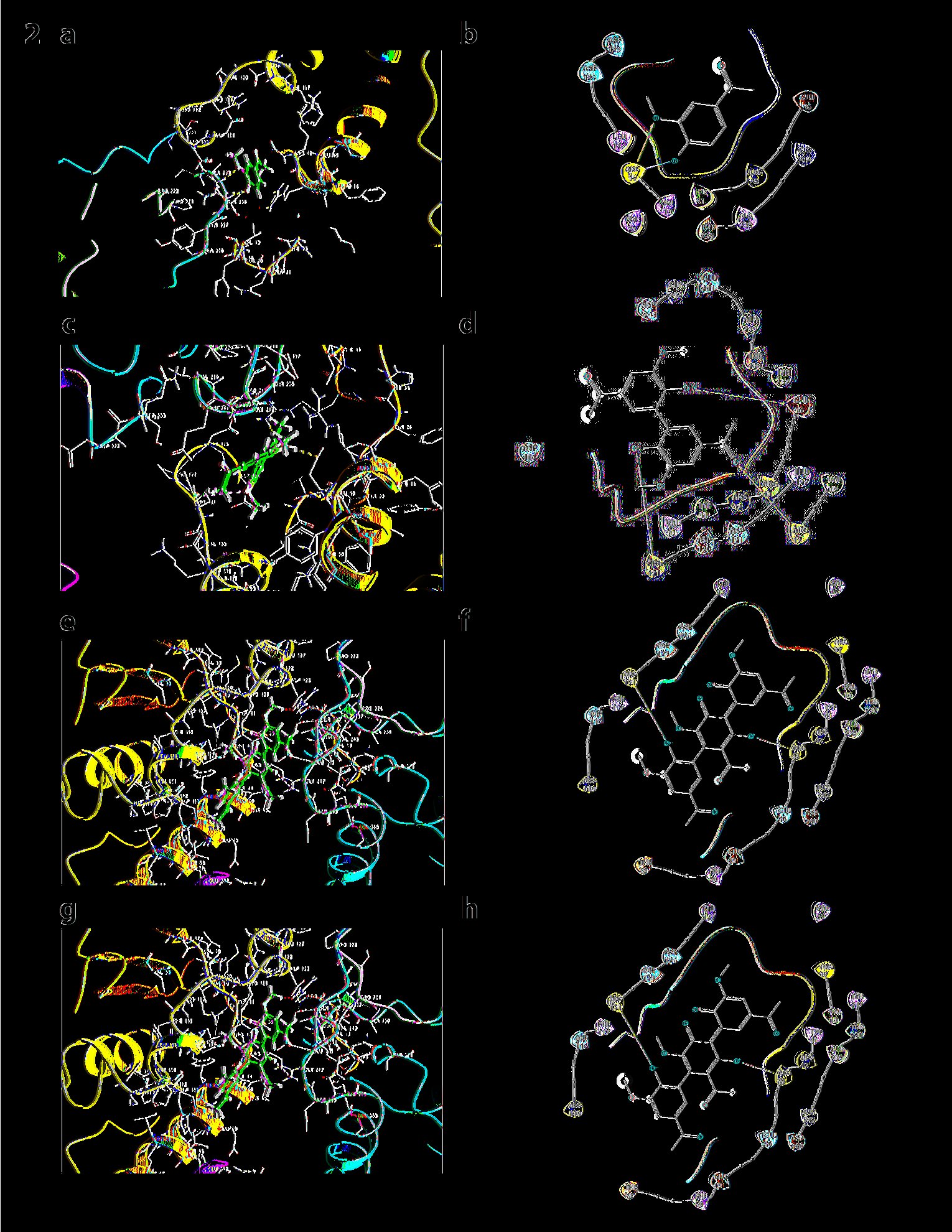
Fig. 2: a) Docking of p47phox with the monomer of apocynin: The monomer of apocynin binds to the PX domain of the p47 subunit. b) Ligand Interaction diagram of the docking of p47phox with the monomer of apocynin. c) Docking of p47phox with apocynin dimer: Dimer of apocynin binds to the AIR domain of the subunit. d) Ligand Interaction diagram of the docking of p47phox with apocynin dimer. e) Docking of p47phox with apocynin trimer (pH7): Apocynin trimer shows docking at AIR domain of p47phox. f) Ligand Interaction diagram of the docking of p47phox with apocynin trimer. g) Docking of p47phox with apocynin trimer (pH8): Trimer at pH 8 shows docking at SH3 domain of p47phox. h) Ligand Interaction diagram of p47phox with apocynin trimer (pH8)
Figures (2e, 2f, 2g, and 2h) depict the docking of trimer at pH 7 and at pH 8 with p47phox complete unit, which are having equal glide scores of -4.99 and Prime energy = -15046.65 Kcal/mol, however, FEB is higher in case of trimer at native pH which is equal to -48.1 kcal/mol and interactions were seen at THR53, LEU127, PHE50, THR238, ARG 121, and GLY 242 at SH3 domain in comparison to trimer at pH8 which shows only one interaction at TYR237 AIR domain with FEB -48.1 Kcal/mol. It can be analyzed from this data that trimer is a better binder than dimer and other forms of Apocynin.
Docking results of apocynin forms with the complete structure of ISF1 of p67phox
Docking results of p67phox isoform1 (NCF2-ISF1) with all forms of apocynin. The monomer of apocynin were docked on TPR domain of p67phox binding (Glide score= -4.283, E model= -30.3, Prime energy= -19682.34 Kcal/mol, FEB= -35.06 Kcal/mol). Dimer again, is seen being docked on TPR domain binding to residues GLU 122 and GLN 169 (Glide score= -6.475, E-model= -49.5, Prime energy= -38.1 Kcal/mol, FEB=-38.1Kcal/mol). Trimer at pH 7 and pH8 have shown no interactions with p67phox isoform1 (NCF2-ISF1) (Fig. 3).
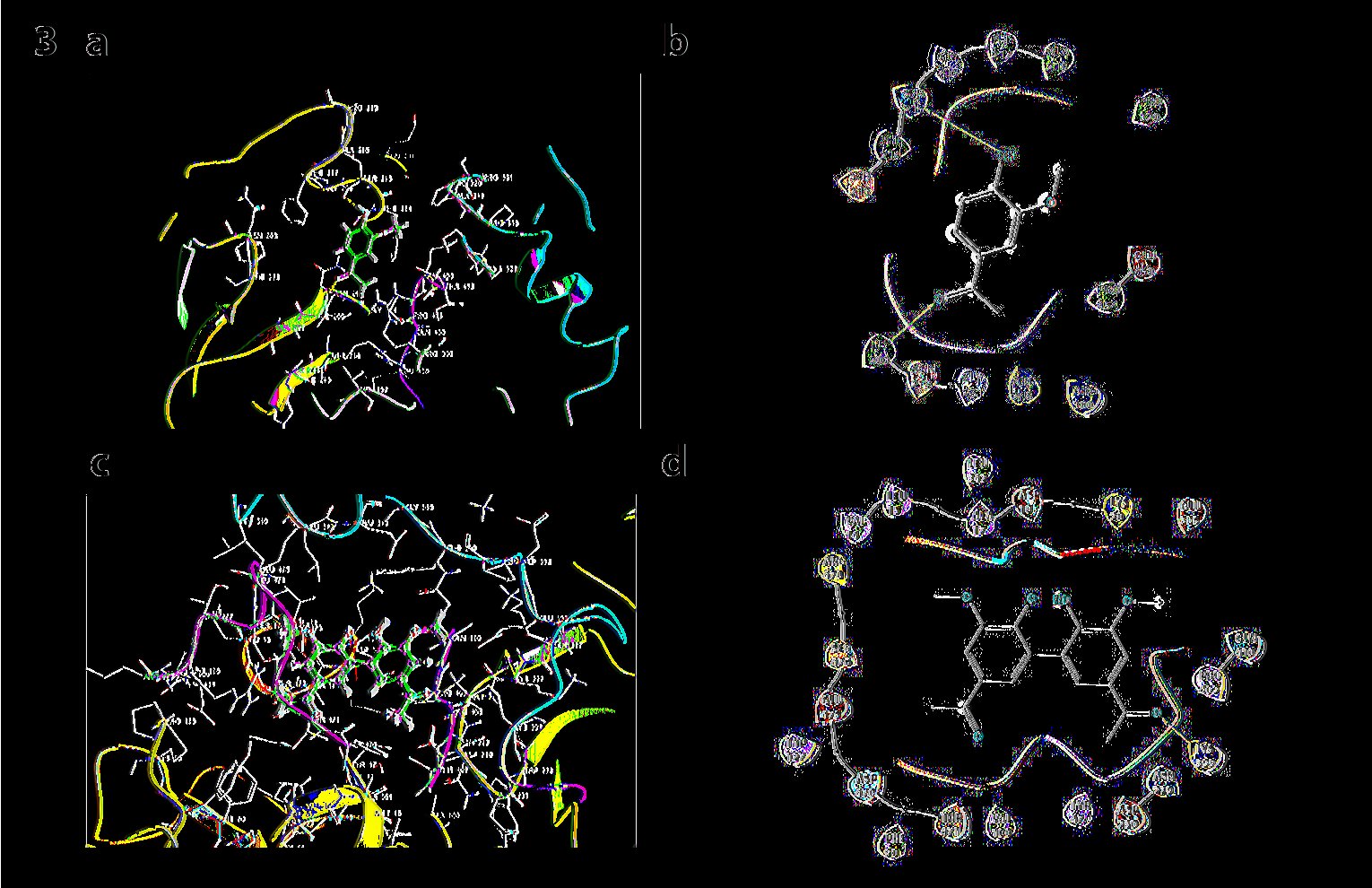
Fig. 3: a) Docking p67phox with monomer: Monomer of apocynin docked on TPR domain. b) Ligand Interaction diagram of the docking p67phox with monomer c) Docking p67phox with dimer: Dimer of apocynin docked on TPR domain. d) Ligand Interaction diagram of the docking p67phox with dimer.
Docking results of apocynin forms with the complete structure of ISF2 of p67phox
Although both ISF2 and ISF1 are isoforms (p67phox) but, each of them shows different interactions with all forms of Apocynin. Figure 4a and 4b show, Monomer gets docked on SH3 domain (Glide score= -3.835, E model=-35.1, Prime energy= -180760.09 Kcal/mol, FEB= -29.29 Kcal/mol). Dimer gets docked on TPR domain (Glide score = -4.176, E model = -69.8, Prime energy = -18767.96 Kcal/mol, FEB = -25.44 Kcal/mol). Dimer has proved out to be a better binder in terms of Glide scores and FEB. Trimer is shown to be docked on SH3 domain (Glide score= -3.347, E model= -59.4, Prime energy=-18758.92-31.19 Kcal/mol, FEB=-31.19). Trimer at pH 8 Trimer is shown to be docked on SH3 domain (Glide score= -3.347, Prime energy=-18758.92-31.19 Kcal/mol, FEB=-31.19).
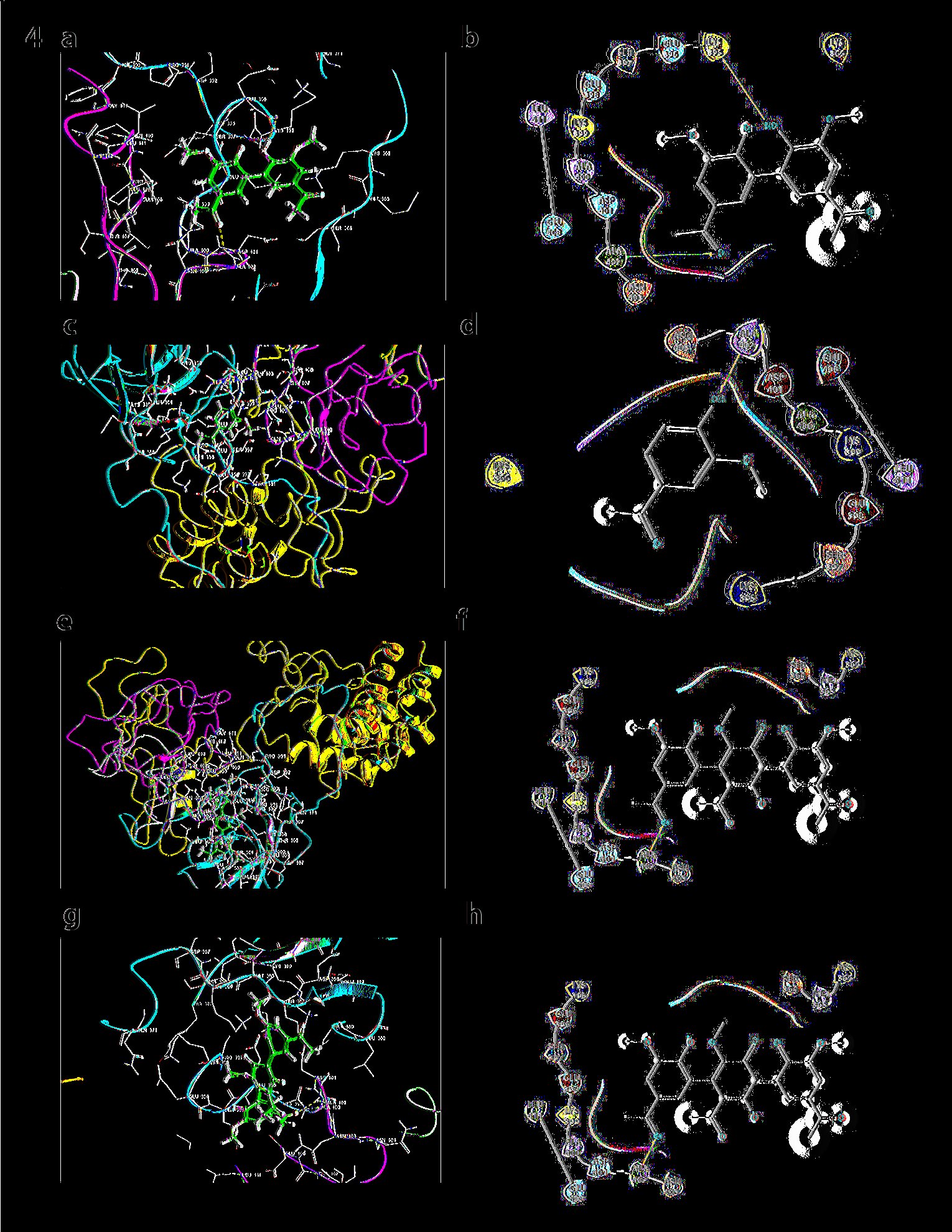
Fig. 4: a) Docking of Monomer with ISF2 of p67phox: Monomer of apocynin docks in the SH3 domain. b) Ligand Interaction diagram of Monomer with ISF2 of p67phox. c) Docking of Dimer with ISF2 of p67phox: Dimer of apocynin docks in the SH3 domain. d) Ligand Interaction diagram of the dimer with ISF2 of p67phox. e) Docking of trimer with ISF2: Trimer of apocynin docked in the SH3 domain. f) Ligand Interaction diagram of ISF2 p67phox. g) Docking of ISF2 with trimmer at pH8: No interactions were observed in this case. h) Ligand Interaction diagram of docking ISF2 with apocynin trimmer at pH8
Docking results of apocynin forms with the complete structure of ISF3 of p67phox
Similarly, the docking results of ISF3 of p67phox have been shown below. As indicated, ISF3 when docked with monomer it interacts in TPR and SH3 domains, respectively. The glide score obtained in case of monomer was -4.356, (Prime energy= -16829.75 Kca/mol, FEB=-30.75) and in the case of dimer, it was -6.151(Prime energy= -16858.71 Kca/mol, FEB=-50.15). In the case of dimer, GLU 154 of ISF3 interacts in the TPR domain. No hydrogen bonds were obtained with trimmer at both the pHs viz 7 or 8. The glide score obtained in case of trimmer at pH7 was -4.136, (Prime energy= -16829.05 Kca/mol, FEB=-33.04) with the glide score at trimmer pH8 was same as trimmer pH7 see Fig. 5).
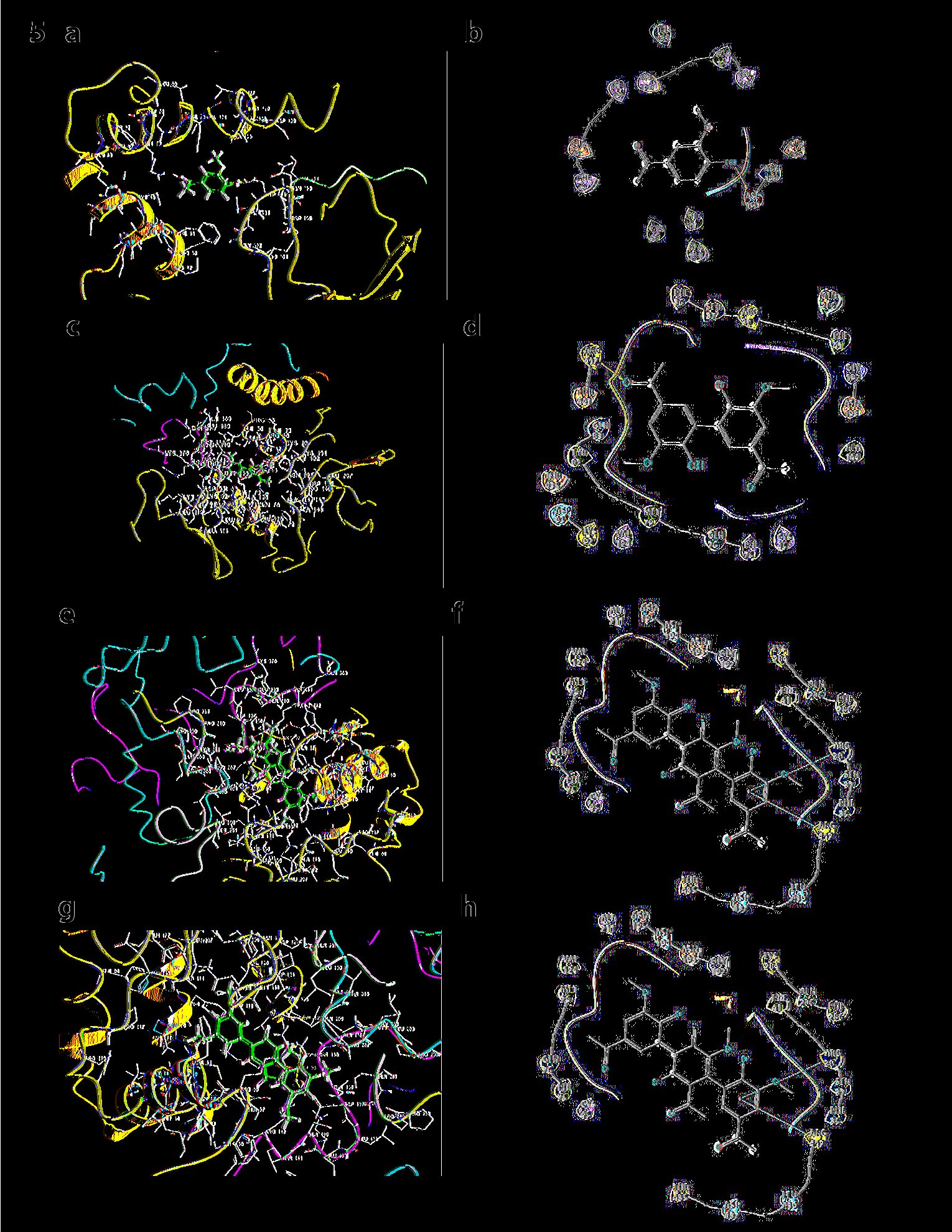
Fig. 5: a) Docking results of ISF3 of p67phox with apocynin monomer. b) Ligand Interaction diagram of docking results of ISF3 of p67phox. c) Docking results of ISF3 of p67phox with apocynin dimer. d) Ligand Interaction diagram of Docking ISF3 of p67phox with apocynin dimer. e) Docking results of ISF3 of p67phox with apocynin trimmer at pH7. f) Ligand Interaction diagram of docking results of apocynin trimmer at pH7 with ISF3 of p67phox. g) Docking results of ISF3 of p67phox with apocynin trimmer at pH8. h) Ligand Interaction diagram of Docking ISF3 of p67phox with apocynin trimmer at pH8
Docking results of apocynin forms with the SH3 domain of p67phox
The SH3 domain of p67phox is important as it interacts with the PRR domain of p47phox; it was docked separately with all forms of apocynin. Monomer of apocynin is seen to dock with Glide score= -2.944, E model = -34.1, Prime energy = -927.71Kcal/mol, FEB = =-30.89Kcal/mol. In case of dimer, it was observed that, dimer is having the lowest FEB as well as glide score (FEB = -39.09Kcal/mol, Glide score = -3.77, E model= -54.6, Prime energy = -951.83 Kcal/mol). Trimer was found to have a Glide score = -2.093, E model = -46.5, Prime energy = -928.82 Kcal/mol, FEB = -31.23 Kcal/mol. In this case, also, dimer has the best FEB in comparison to others. In all the cases these forms were docked into the same C-terminal SH3 domain. In the case of trimer at pH8, the trimer is seen to interact with THR332 (Fig. 6).
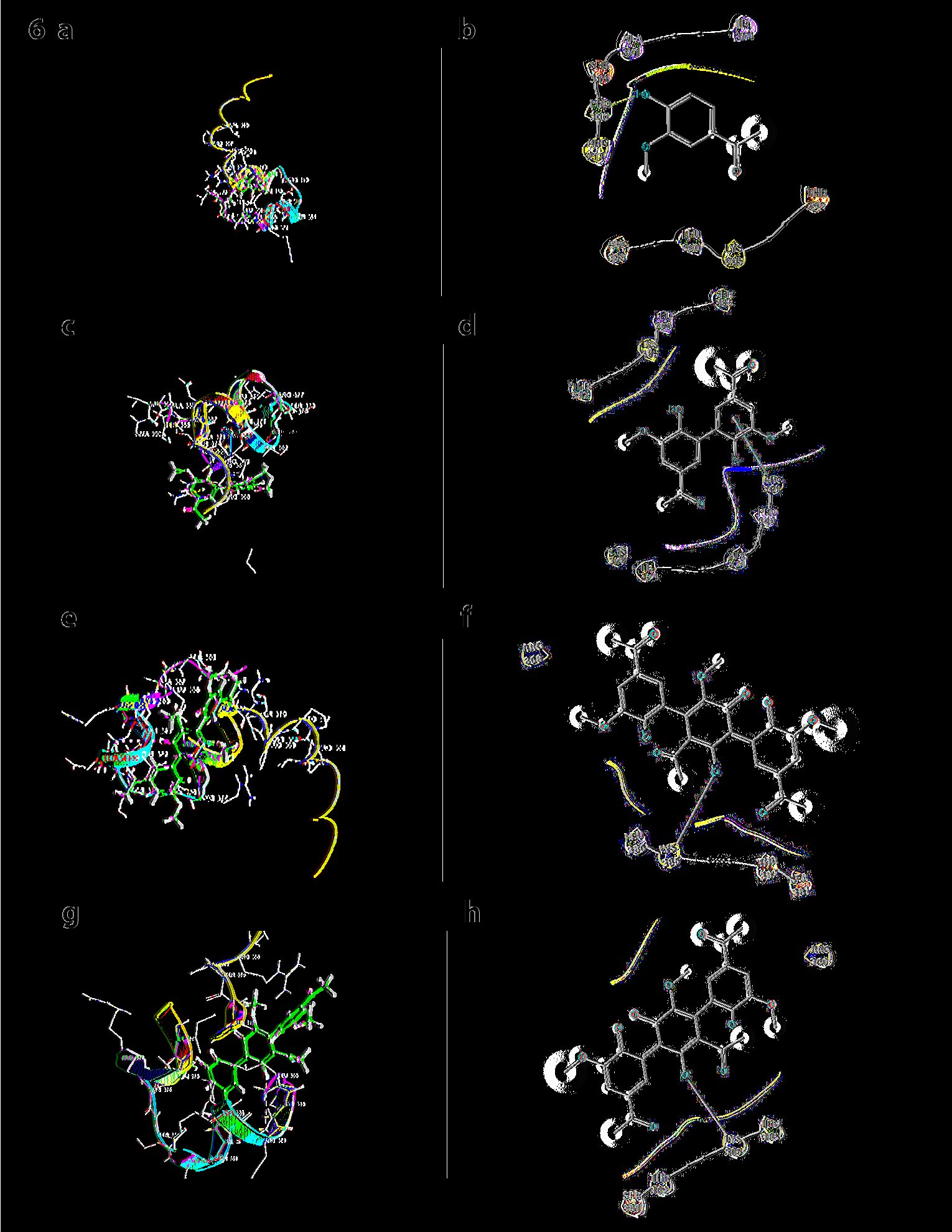
Fig. 6: a) Docking SH3 domain of p67phox with monomer: Monomer of apocynin. b) Ligand Interaction diagram of the SH3 domain of p67phox with monomer. c) Docking SH3 domain of p67phox with dimer: Dimer of apocynin. d) Ligand Interaction diagram of the SH3 domain of p67phox with dimer. e) Docking SH3 domain of p67phox with trimmer: Trimer of apocynin. f) Ligand Interaction diagram of the SH3 domain of p67phox. g) Docking SH3 domain of p67phox with trimmer at pH8: Monomer interacts with SH3 domain. h) Ligand Interaction diagram of the SH3 domain of p67phox with trimmer
Table 1 shows the glide scores and the prime energies (in brackets) of docking that are indicative of the affinity of a particular ligand towards the target site or protein in question.
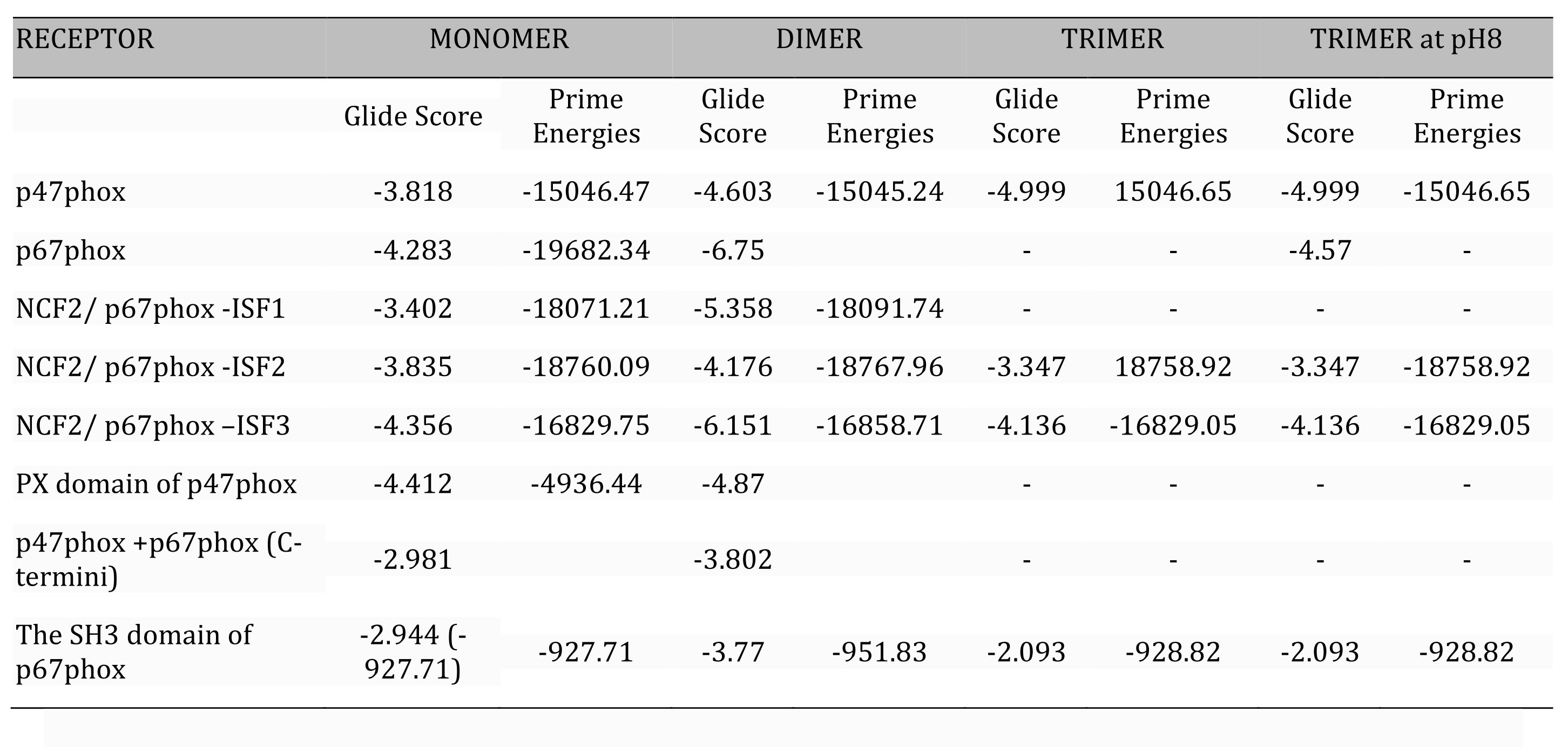
Table 1: Comparative table of glide score* and the prime energies values of three forms of apocynin docked on subunits of NOX. *(ΔG Kcal/mol), Prime Energy given in brackets
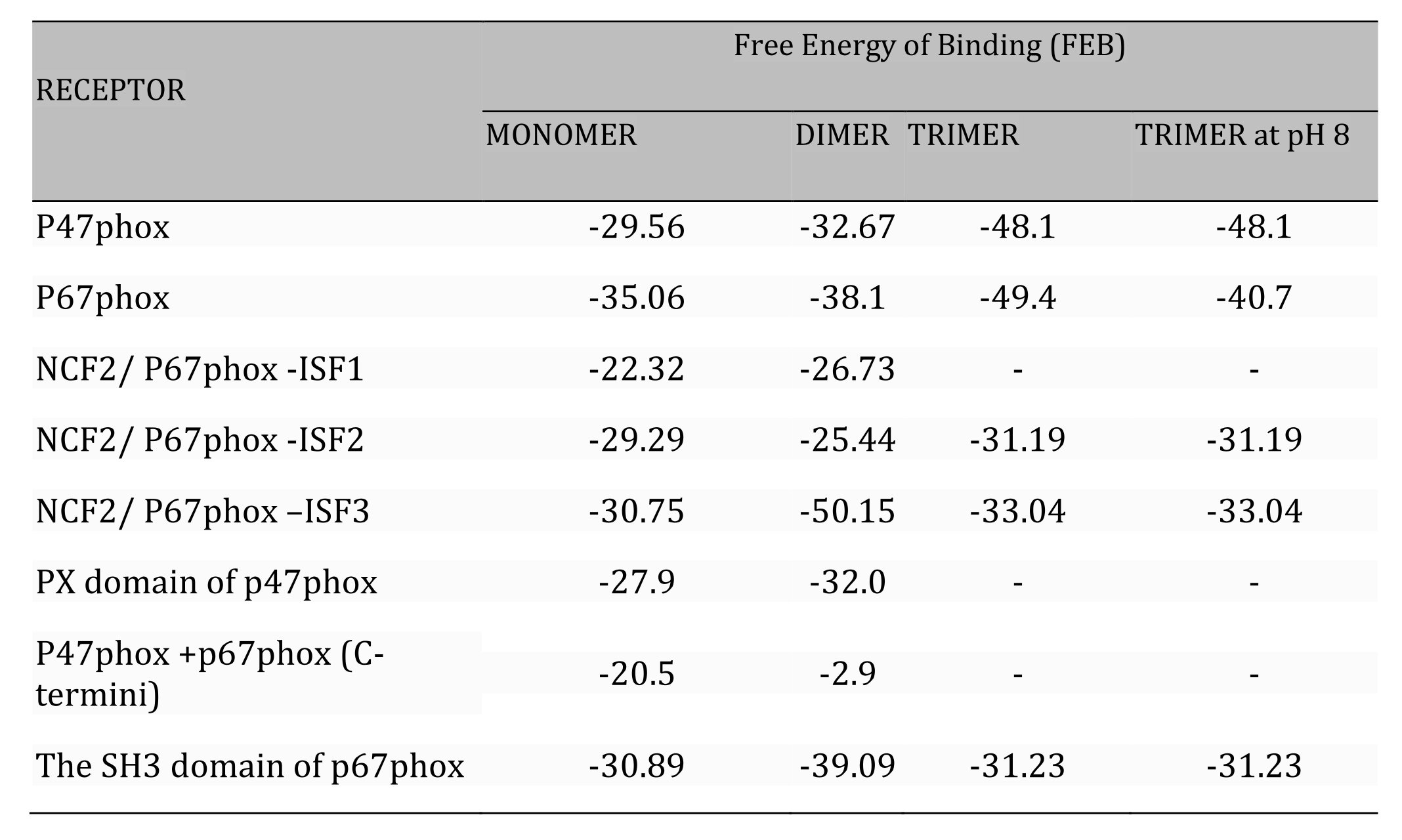
Table 2: Comparative table of Free Energy of Binding* (FEB) of three forms of apocynin docked on subunits of NOX
Molecular Dynamics Simulation Analysis
Molecular dynamics (MD) simulation is performed in in silico drug discovery to assess the dynamic stability and intermolecular interactions between the protein and ligand in a docked complex with respect to time. In MD simulation, root mean square deviation (RMSD) is used to analyze the stability of docked protein-ligand complex, whereas the root mean square fluctuation (RMSF) is used to analyses the internal fluctuations of amino acid residues in protein and atoms in the ligands. In addition, protein-ligand interaction mapping analysis in MD simulation is used to calculate intermolecular interactions in the docked complex with respect to time during the simulation to assess the stability of protein with the docked ligands. The RMSD and RMSF values along with the protein-ligand contact profiles are extracted from the respective 100 ns simulation trajectories as shown in Figure 7.
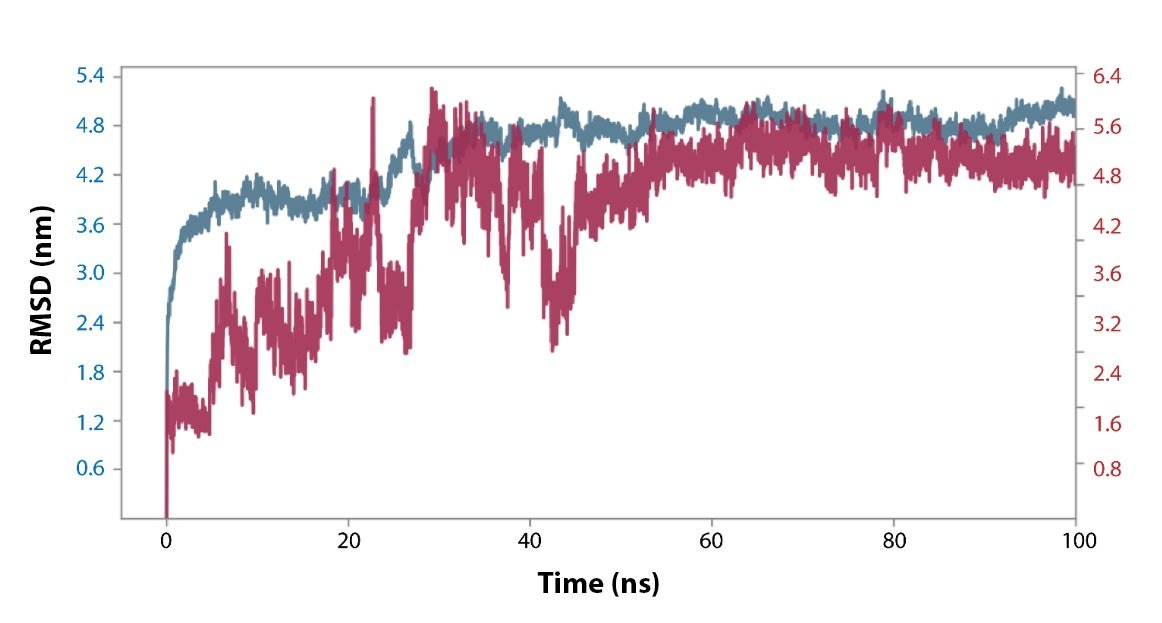
Fig. 7: RMSD plot of the Apo-dimer-ISF3 protein-ligand docked complex were extracted from 100 ns MD simulation trajectories of respective docked complexes.
RMSD and RMSF Analysis. The RMSD and RMSF of protein and ligand in the Apo-dimer-ISF3 docked complex were analyzed to were analyzed with respect to the initial pose as a reference frame. In the docked complex, the Apo-dimer is showing high deviation with the RMSF value of ~4.0 Å till 30 to 35 ns and later it deviated beyond 4.0 Å and equilibrated at ~5.0 Å. The ligand ISF3 showed continuous fluctuation till 50 ns followed by the equilibrium attained at ~5.0 Å. The RMSD values of protein and ligand were supported by the respective RMSF values of amino acid residues in Apo-dimer (>4.0 Å) and atoms in the ISF3 ligand (4.0 0197), where the internal fluctuation was higher.
Protein-ligand Interaction Profiling. The Apo-dimer-ISF3 docked complex were further considered for the protein-ligand interaction profiling against the reference docked complex concerning the hydrogen bonding, hydrophobic interactions, ionic interactions, and water bridge formation during the simulation time period during 100 ns MD simulation interval as shown in Figure 8.
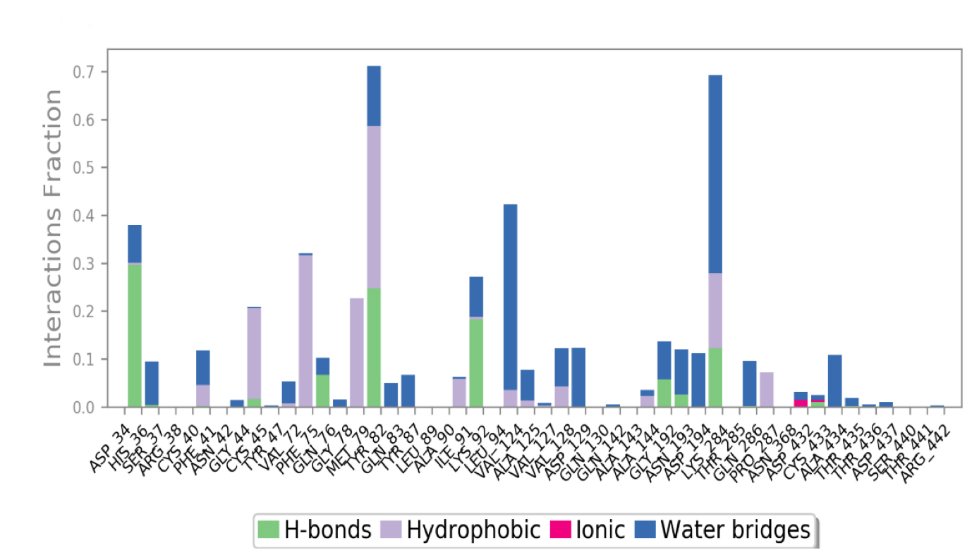
Fig. 8: Protein-ligand interactions mapping for Apo-dimer-ISF3 protein-ligand docked complex were extracted from 100 ns MD simulation trajectories of respective docked complexes.
In the docked complex, His36, Tyr82, and Lys92 showed the formation of hydrogen bonding for more than 20% of the total simulation time. The hydrophobic interaction was formed by residues Cys45, and Met79 for more than 20% whereas, by residues Phe75, and Tyr82 for more than 30% of the simulation interval. Moreover, few residues also formed water bridge for shorter period during the simulation interval.
Discussion
NOX, the enzyme complex which is linked to many diseases of inflammation is being used as a target to inhibit the generation of superoxide radicals thereby reducing inflammation, and apoptosis [42]. Much research has been done on natural molecules and synthetic compounds to inhibit NOX. Apocynin has thus far been found to be one of the most effective inhibitors and has been proven to be a safer molecule than the rest, being a natural compound [43-45]. It has been reported that apocynin itself doesn’t show any activity unless it is dimerized in presence of peroxidases [46]. Dimer has also been shown to inhibit the translocation of p47phox and p67phox and found to have a role to inhibit Rac and Rac related homologues [47]. In addition, not only dimeric but trimeric forms of apocynin have also been reported for its biological activity [1, 4, 14]. Therefore, based on literature, we had selected three forms of Apocynin: monomer, dimer, and trimer, to study its interactions with the enzyme NOX. It has been investigated whether apocynin is directly involved to interact with the enzyme. These three forms of apocynin were mainly docked on three subunits of NOX enzyme namely p47phox, p67phox and Rac 1, and on the complex of p47phox and p47phox. Out of all three forms, trimer was selected to be docked at two different pH levels, based on the studies in the literature, one being at 7±2 and other at 8±2.
As reported in the introduction, reasons for selecting specific structures discussed in this study were: p67phox & p47phox. Apocynin is reported to inhibit translocation of p67phox and SH3 domain is mainly responsible for binding to p47phox so if, apocynin interacts with either or both of them it may affect their recruitment at the membrane [4, 10, 16, 17, 48].
PX Domain of p47phox: PX domain of p47phox is responsible for primary activation and translocation of p47phox [49]. As it was reported that apocynin inhibits translocation of p47phox [16] so, this domain was selected to check if this interaction is responsible for the suggested findings.
p47phox & p67phox Complex: p47phox and p67phox exist as a complex in cytosol [10, 50]. We selected this structure (PDB ID-1K4U) for viewing interactions with the three forms of apocynin to check whether any form of apocynin can stabilize this complex in the cytosol itself and further inhibit their translocation.
Rac & Rac Homologues: Apocynin was reported to play an inhibitory role over Rac and Rac homologues [1, 51, 52] to which p47phox and p67phox are ultimately recruited before complete activation of the enzyme. It was chosen for docking to check whether the inhibition of the enzyme takes place at this stage also. The results shown that there is no prominent interaction between all the forms of Apocynin with Rac and Rac homologues.
Rigid docking of dimer on the protein complex of C-terminal region of p47phox showed glide score of -4.83. It has been reported that, if SER379 is phosphorylated then, NOX cannot aggregate together and therefore becomes inactive [15, 16]. Monomer also docked on the same cavity but on a different residue SER381. This spanning region of residues from 359-390 is the PRR domain region of p47phox, which is responsible for translocation of p67phox to membrane region as PRR domain, binds to the SH3 domain on p67phox [53]. Whereas, when p47phox is absent from the complex p47phox- p67phox, we observed that three forms of apocynin could readily bind to p67phox with a very good FEB. So, it was indicative that p67phox could also be inhibited directly by Dimeric and Trimeric forms of apocynin. In the SH3 domain of p67phox, LYS502 has been found common in binding with all three forms of apocynin so, there may be an activity involved with this residue. Also, it was seen that the trimer didn’t bind on p67phox when it is complexed with p47phox but showed activity when free SH3 domain of p67phox was present. Might be p47phox hides the active sites of p67phox or the same site is involved in binding with p47phox itself. It seems that, in both the cases of dimer and trimer, p47phox might be the main target. When only the PX domain of p47phox was used for docking with the three forms of apocynin, only monomer and dimer showed binding with dimer being the efficient binder as compared to monomer, showing hydrogen bond interactions with maximum number of residues.
Further, when three forms of apocynin were docked on p67phox complete subunit, once again trimer obtained the highest FEB. It can be inferred that trimer can readily bind with the p67phox complete subunit as compared to other three forms of apocynin and proved out to be the best binder amongst all three forms of apocynin. Also, trimer shows interactions with maximum number of residues, as was the case with p47phox complete subunit. Trimmer at pH8 has the lowest FEB hence, doesn’t show effective binding. When three forms of Apocynin docking with different isoforms of p67phox were compared it was found that, among all the isoforms except ISF-2, trimer showed the highest FEB, keeping it synonymous with the p67phox complete subunit results. In the case of ISF-2, dimer showed the best FEB values amongst all three forms of apocynin. In case of ISF-3 docking with trimmer didn’t yield any hydrogen bond formation but, FEB of the complex was sufficiently large suggesting the trimer to dominate in this case as well, compared to all three forms of apocynin. Docking on NCF1-ISF1 shows that the three forms of apocynin have affinity of binding in the pocket region of PX domain and SH3 domain. This is an indicative of the reported inhibitory activity of apocynin even in isoforms of p67phox. In general, the glide scores and Free Energy of binding (FEB) of trimer were found to be better than dimer and monomer in most of the cases when docked on active domains of the subunits/subunit complex. According to MDS studies, the results have been validated that the Apocynin trimer molecule have a better stability and interaction with the enzyme complex which in turn shows to be more effective binder and may be a potential inhibitor for various subunits of this enzyme.
Conclusion
After conducting this study, it can be predicted that dimer and trimer of apocynin, both might have a significant role in interaction with NOX cytosolic subunits. The interaction of dimer on SER379 in p47phox- p67phox complex may have a significant role as this residue is thought to be of importance as when phosphorylated it can bring about total deactivation of NOX enzyme. In addition, the Rac-GDP complex also showed significant interactions with forms of apocynin hence suggesting a role in cellular signaling as reported in literature [16]. Interactions with trimer has also shown a better glide score and FEB when docked to p47phox and p67phox complete structure and its isoforms, and therefore it can be said that a trimer may as well play a role in inhibition of NOX. In case of p67phox isoforms as well, all three forms of apocynin turned out to be binders with high FEB, except for ISF-2 and trimmer combination. In the rest of the cases, the trimeric form of apocynin proved out to be the most effective binder amongst all three forms of apocynin. The study has been also validated by MDS revealing its better stability and interaction.
Acknowledgements
The authors would like to acknowledge the Department of Biotechnology, Jaypee Institute of Information Technology, Noida, India for providing the technical and infrastructural support to carry out this study. All the authors have contributed equally. This research received no external funding. The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors have no conflicts of interest to declare.
References
| 1 | Bedard K, Krause KH: The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245-313.
https://doi.org/10.1152/physrev.00044.2005 |
| 2 | Drummond GR, Selemidis S, Griendling KK, Sobey CG: Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 2011;10:453-471.
https://doi.org/10.1038/nrd3403 |
| 3 | Heyworth PG, Cross AR, Curnutte JT: Chronic granulomatous disease. Curr Opin Immunol 2003;15:578-584.
https://doi.org/10.1016/S0952-7915(03)00109-2 |
| 4 | Yu J, Weïwer M, Linhardt RJ, Dordick JS: The role of the methoxyphenol apocynin, a vascular NADPH oxidase inhibitor, as a chemopreventative agent in the potential treatment of cardiovascular diseases. Curr Vasc Pharmacol 2008;6:204-217.
https://doi.org/10.2174/157016108784911984 |
| 5 | Sumimoto H: Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J 2008;275:3249-3277.
https://doi.org/10.1111/j.1742-4658.2008.06488.x |
| 6 | Batchuluun B, Pinkosky SL, Steinberg GR: Lipogenesis inhibitors: therapeutic opportunities and challenges. Nat Rev Drug Discov 2022;21:283-305.
https://doi.org/10.1038/s41573-021-00367-2 |
| 7 | Cifuentes-Pagano E, Meijles DN, Pagano PJ: The quest for selective nox inhibitors and therapeutics: challenges, triumphs and pitfalls. Antioxid Redox Signal 2014;20:2741-2754.
https://doi.org/10.1089/ars.2013.5620 |
| 8 | Cifuentes-Pagano E, Csanyi G, Pagano PJ: NADPH oxidase inhibitors: a decade of discovery from Nox2ds to HTS. Cell Mol Life Sci 2012;69:2315-2325.
https://doi.org/10.1007/s00018-012-1009-2 |
| 9 | Kumar Tiwari A, Gandhi V, Agarwal S, et al.: In-silico validation of Apocynin and NADPH Oxidase (NOX) enzyme for inhibiting ROS injuries. Mater Today 2023;80: 2375-2377.
https://doi.org/10.1016/j.matpr.2021.06.361 |
| 10 | Sumimoto H, Kage Y, Nunoi H, et al.: Role of Src homology 3 domains in assembly and activation of the phagocyte NADPH oxidase. PNAS 1994;91:5345-5349.
https://doi.org/10.1073/pnas.91.12.5345 |
| 11 | Panday A, Sahoo MK, Osorio D, Batra S: NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol 2015;12:5-23.
https://doi.org/10.1038/cmi.2014.89 |
| 12 | Leto TL, Morand S, Hurt D, Ueyama T: Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid Redox Signal 2009;11:2607-2619.
https://doi.org/10.1089/ars.2009.2637 |
| 13 | Groemping Y, Rittinger K: Activation and assembly of the NADPH oxidase: a structural perspective. Biochem 2005;386:401-416
https://doi.org/10.1042/BJ20041835 |
| 14 | Leto TL, Adams AG, de Mendez I: Assembly of the phagocyte NADPH oxidase: binding of Src homology 3 domains to proline-rich targets. PNAS 1994;91:10650-10654.
https://doi.org/10.1073/pnas.91.22.10650 |
| 15 | Kawahara T, Quinn MT, Lambeth JD: Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol Biol 2007;7:109.
https://doi.org/10.1186/1471-2148-7-109 |
| 16 | Stefanska J, Pawliczak R: Apocynin: molecular aptitudes. Mediators Inflamm 2008;2008:106507.
https://doi.org/10.1155/2008/106507 |
| 17 | Touyz RM: Apocynin, NADPH Oxidase, and Vascular Cells. Hypertens 2008;51:172-174.
https://doi.org/10.1161/HYPERTENSIONAHA.107.103200 |
| 18 | Torres PHM, Sodero ACR, Jofily P, Silva-Jr FP: Key Topics in Molecular Docking for Drug Design. Int J Mol Sci 2019;20.
https://doi.org/10.3390/ijms20184574 |
| 19 | Ramachandran B, Kesavan S, Rajkumar T: Molecular modeling and docking of small molecule inhibitors against NEK2. Bioinformation 2016;12:62-68.
https://doi.org/10.6026/97320630012062 |
| 20 | Hetényi C, van der Spoel D: Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett 2006;580:1447-1450.
https://doi.org/10.1016/j.febslet.2006.01.074 |
| 21 | Friesner RA, Murphy RB, Repasky MP, et al.: Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J Med Chem 2006;49:6177-6196.
https://doi.org/10.1021/jm051256o |
| 22 | Lipinski CA, Lombardo F, Dominy BW, Feeney PJ: Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001;46:3-26.
|
| 23 | Hiroaki H, Ago T, Ito T, Sumimoto H, Kohda D: Solution structure of the PX domain, a target of the SH3 domain. Nat Struct Biol 2001;8:526-530.
https://doi.org/10.1038/88591 |
| 24 | Kami K, Takeya R, Sumimoto H, Kohda D: Diverse recognition of non-PxxP peptide ligands by the SH3 domains from p67phox, Grb2 and Pex13p. EMBO J 2002;21:4268-4276.
https://doi.org/10.1093/emboj/cdf428 |
| 25 | Rose PW, Prlić A, Altunkaya A, et al.: The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res 2017;45(D1):D271-d281.
|
| 26 | Zheng W, Zhang C, Bell EW, Zhang Y: I-TASSER gateway: A protein structure and function prediction server powered by XSEDE. Future generations computer systems. FGCS 2019;99:73-85.
https://doi.org/10.1016/j.future.2019.04.011 |
| 27 | Feng P, Liang JY, Li TL, et al.: Zinc induces mitochondria apoptogenesis in prostate cells. Mol Urol 2000;4:31-36.
|
| 28 | Bhachoo J, Beuming T: Investigating Protein-Peptide Interactions Using the Schrödinger Computational Suite. In: Schueler-Furman O, London N, eds. Modeling Peptide-Protein Interactions: Methods and Protocols. Springer New York; 2017:235-254.
https://doi.org/10.1007/978-1-4939-6798-8_14 |
| 29 | LigPrep, version 2.5. Schrödinger, LLC,0 NY. 2011.
|
| 30 | Halgren TA, Murphy RB, Friesner RA, et al.: Glide: A New Approach for Rapid, Accurate Docking and Scoring. Enrichment Factors in Database Screening. J Chem Med 2004;47:1750-1759.
https://doi.org/10.1021/jm030644s |
| 31 | Friesner RA, Banks JL, Murphy RB, et al.: Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1 Method and Assessment of Docking Accuracy. J Med Chem 2004;47:1739-1749.
https://doi.org/10.1021/jm0306430 |
| 32 | Park MS, Gao C, Stern HA: Estimating binding affinities by docking/scoring methods using variable protonation states. Proteins 2011;79:304-314.
https://doi.org/10.1002/prot.22883 |
| 33 | Genheden S, Ryde U: The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 2015;10:449-461.
https://doi.org/10.1517/17460441.2015.1032936 |
| 34 | Kutzner C, Páll S, Fechner M, Esztermann A, de Groot BL, Grubmüller H: More bang for your buck: Improved use of GPU nodes for GROMACS 2018. J Comput Chem 2019;40:2418-2431.
https://doi.org/10.1002/jcc.26011 |
| 35 | Huang J, Rauscher S, Nawrocki G, et al.: CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 2017;14:71-73.
https://doi.org/10.1038/nmeth.4067 |
| 36 | Vanommeslaeghe K, Raman EP, MacKerell AD, Jr: Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J Chem Inf Model 2012;52:3155-3168.
https://doi.org/10.1021/ci3003649 |
| 37 | Tom Darden DY, Lee Pedersen: Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J Chem Phys 1993;98.
https://doi.org/10.1063/1.464397 |
| 38 | Bussi G, Donadio D, Parrinello M: Canonical sampling through velocity rescaling. J Chem Phys 2007;126:014101.
https://doi.org/10.1063/1.2408420 |
| 39 | Parrinello M, Rahman A: Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys Rev Lett 1980;45:1196-1199.
https://doi.org/10.1103/PhysRevLett.45.1196 |
| 40 | Singh R, Meena NK, Das T, Sharma RD, Prakash A, Lynn AM: Delineating the conformational dynamics of intermediate structures on the unfolding pathway of β-lactoglobulin in aqueous urea and dimethyl sulfoxide. J Biomol Struct Dyn 2020;38:5027-5036.
https://doi.org/10.1080/07391102.2019.1695669 |
| 41 | Prakash A, Dixit G, Meena NK, et al.: Elucidation of stable intermediates in urea-induced unfolding pathway of human carbonic anhydrase IX. J Biomol Struct Dyn 2018;36:2391-2406.
https://doi.org/10.1080/07391102.2017.1355847 |
| 42 | Tarafdar A, Pula G: The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int J Mol Sci 2018;19.
https://doi.org/10.3390/ijms19123824 |
| 43 | Zimmerman HJ: Hepatotoxicity. Dis Mon 1993;39:675-787.
https://doi.org/10.1016/0011-5029(93)90015-U |
| 44 | Lee WM. Drug-induced hepatotoxicity. N Engl J Med 1995;333:1118-1127.
https://doi.org/10.1056/NEJM199510263331706 |
| 45 | Petrônio MS, Zeraik ML, Fonseca LMd, Ximenes VF: Apocynin: Chemical and Biophysical Properties of a NADPH Oxidase Inhibitor. Molecules 2013;18:2821-2839.
https://doi.org/10.3390/molecules18032821 |
| 46 | Ximenes VF, Kanegae MP, Rissato SR, Galhiane MS: The oxidation of apocynin catalyzed by myeloperoxidase: proposal for NADPH oxidase inhibition. Arch Biochem 2007;457:134-141.
https://doi.org/10.1016/j.abb.2006.11.010 |
| 47 | Van den Worm E, Beukelman CJ, Van den Berg AJJ, Kroes BH, Labadie RP, Van Dijk H: Effects of methoxylation of apocynin and analogs on the inhibition of reactive oxygen species production by stimulated human neutrophils. Eur J Pharmacol 2001;433:225-230.
https://doi.org/10.1016/S0014-2999(01)01516-3 |
| 48 | Schröder K, Weissmann N, Brandes RP: Organizers and activators: Cytosolic Nox proteins impacting on vascular function. Free Radic Biol Med 2017;109:22-32.
https://doi.org/10.1016/j.freeradbiomed.2017.03.017 |
| 49 | Karathanassis D, Stahelin RV, Bravo J, et al.: Binding of the PX domain of p47(phox) to phosphatidylinositol 3, 4-bisphosphate and phosphatidic acid is masked by an intramolecular interaction. EMBO J 2002;21:5057-5068.
https://doi.org/10.1093/emboj/cdf519 |
| 50 | Iyer SS, Pearson DW, Nauseef WM, Clark RA: Evidence for a readily dissociable complex of p47phox and p67phox in cytosol of unstimulated human neutrophils. J Biol Chem 1994;269:22405-22411.
https://doi.org/10.1016/S0021-9258(17)31804-5 |
| 51 | Ueyama T, Geiszt M, Leto TL: Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Mol Cell Biol 2006;26:2160-2174.
https://doi.org/10.1128/MCB.26.6.2160-2174.2006 |
| 52 | Ueno N, Takeya R, Miyano K, Kikuchi H, Sumimoto H: The NADPH oxidase Nox3 constitutively produces superoxide in a p22phox-dependent manner: its regulation by oxidase organizers and activators. J Biol Chem2005;280:23328-23339
https://doi.org/10.1074/jbc.M414548200 |
| 53 | Taura M, Miyano K, Minakami R, Kamakura S, Takeya R, Sumimoto H: A region N-terminal to the tandem SH3 domain of p47phox plays a crucial role in the activation of the phagocyte NADPH oxidase. Biochem 2009;419:329-338.
https://doi.org/10.1042/BJ20082028 |