Autophagy Inhibition Increased Sensitivity of Pancreatic Cancer Cells to Carbon Ion Radiotherapy
Keywords
Abstract
Background/Aims:
Pancreatic cancer has the poorest survival rate among all cancer types. Therefore, it is essential to develop an effective treatment strategy for this cancer.Methods:
We performed carbon ion radiotherapy (CIRT) in human pancreatic cancer cell lines and analyzed their survival, apoptosis, necrosis, and autophagy. To investigate the role of CIRT-induced autophagy, autophagy inhibitors were added to cells prior to CIRT. To evaluate tumor formation, we inoculated CIRT-treated murine pancreatic cancer cells on the flank of syngeneic mice and measured tumor weight. We immunohistochemically measured autophagy levels in surgical sections from patients with pancreatic cancer who received neoadjuvant chemotherapy (NAC) plus CIRT or NAC alone.Results:
CIRT reduced the survival fraction of pancreatic cancer cells and induced apoptotic and necrotic alterations, along with autophagy. Preincubation with an autophagy inhibitor accelerated cell death. Mice inoculated with control pancreatic cancer cells developed tumors, while those inoculated with CIRT/autophagy inhibitor-treated cells showed significant evasion. Surgical specimens of NAC-treated patients expressed autophagy comparable to control patients, while those in the NAC plus CIRT group expressed little autophagy and nuclear staining.Conclusion:
CIRT effectively killed the pancreatic cancer cells by inhibiting their autophagy-inducing abilities.Introduction
Pancreatic cancer is typically incurable and characterized by having the poorest survival rate among all cancer types [1, 2]. The International Agency for Research on Cancer estimated approximately 10.0 million cancer-related deaths worldwide in 2020, and that pancreatic cancer was the seventh leading cause of cancer-related deaths (466, 000: 4.7% of all cancer-related deaths worldwide in 2020) in both sexes [1]. Pancreatic cancer is difficult to diagnose at an early stage owing to a lack of specific symptoms. In fact, by the time of diagnosis, pancreatic cancer often metastasizes to the surrounding and/or distant organs and spreads to the drainage lymph nodes [2]. Because of this, many pancreatic cancers are diagnosed as unresectable, with only 20% being resectable. A combination of surgery and chemotherapy has become the standard treatment strategy for pancreatic cancer. However, the prognosis of this disease remains poor, and the general five-year survival rate is only 10% [2]. Neoadjuvant therapy consisting of chemotherapy and/or radiotherapy is used for non-metastatic but unresectable locally advanced pancreatic cancer (LAPC) [3]. Some clinical trials using photon radiation therapy have been inconclusive in patients with LAPC [4-6]. Thus, a novel and effective treatment strategy for pancreatic cancer is required.
Approximately 90% of pancreatic cancers possess point mutations in the KRAS oncogene [2, 7]. RAS proteins generated from mutant KRAS constitutively activate their downstream signaling pathways, such as those involved in the growth, proliferation, and survival of cancer cells [8]. This may be responsible for the aggressive growth of pancreatic cancer. However, preclinical trials of inhibitors of the downstream pathways of mutant RAS proteins have failed to achieve satisfactory results [9, 10].
Carbon ion radiation therapy (CIRT) for cancer started from 1994 at the National Institute of Radiological Science (currently known as National Institutes for Quantum Science and Technology, Japan) [3]. Because of their pin-point dose distribution owing to the Bragg peak, carbon ion beams provide cancer cells with a peak dose radiation without exerting any adverse effects on normal tissues in the vicinity [11]. Moreover, carbon ion beams have 2–3 folds higher relative biological effectiveness (RBE) than photon beams, such as X-rays and g-rays, as carbon particles are 12-fold heavier and have higher linear energy transfer (LET) than proton beams [12]. A higher LET can induce double-strand breaks in DNA. Recent clinical trials have demonstrated the protective effects of CIRT on patients with pancreatic cancer [13, 14].
Ionizing radiation, including CIRT, generates multiple cellular responses, such as generation of reactive oxygen species (ROS) [15], induction of the unfolded protein response [16], apoptosis [17], metabolic imbalance, and DNA damage, which are responsible for its anticancer activity. However, ionizing radiation can have pro-cancer effects, while also facilitating activation of autophagy [18-21]. Some studies have revealed that autophagy aids in cell survival [18-21] and maintains energy homeostasis [22]. This prompted us to investigate whether the inhibition of autophagy may improve the therapeutic effects of CIRT on pancreatic cancer.
Materials and Methods
Cell culture and reagents
Human pancreatic cancer cell lines (MIA PaCa-2, BxPC3, and PANC 1) [23] were purchased from the American Type Culture Collection (Manassas, Virginia, USA). These cell lines were maintained in the Roswell Park Memorial Institute 1640 medium purchased from Thermo Fisher Scientific K.K. (Tokyo, Japan) supplemented with 10% fetal bovine serum and incubated at 37 °C and 5% CO2. Pan02, established from the pancreatic cancer cells of male C57BL/6 mice treated with the cancer-promoting chemical 3-methylcholanthrene [24], was purchased from the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute (Frederick, MD, USA). Pan02 cells were maintained in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific K.K., Tokyo, Japan) with 10% fetal bovine serum and incubated at 37 °C and 5% CO2. Hydroxychloroquine (sulfate) (HCQ) was purchased from CAYMAN CHEMICAL (MI, USA). The pro-apoptotic reagent, paclitaxel, was purchased from Selleck Chemicals (TX, USA). Antibodies against phosphorylated p44/p42 MAPK (T202/Y204), p44/p42 (Erk1/2), poly ADP-ribose polymerase (PARP), Caspase-3, and microtubule-associated proteins 1A/1B light chain 3B (LC3B) were purchased from Cell Signaling Technology (MA, USA). Anti-LC3 antibody (clone: LC3 No.6) was purchased from Cosmo Bio (Tokyo, Japan). Monoclonal antibodies against p62 (SQSTM1) were purchased from MEDICAL & BIOLOGICAL LABORATORIES (Tokyo, Japan). The anti-β-actin antibody was purchased from Sigma-Aldrich (MO, USA).
In vitro carbon ion irradiation
For in vitro carbon ion irradiation, cells in T25 culture flasks (IWAKI, Japan) were irradiated with 100 MeV/u horizontal carbon ion beams at the Osaka Heavy Ion Therapy Center (Osaka, Japan). The cell culture surface was placed at about 2 mm before Braggs peak, where the LET was approximately 80 KeV/mm. In some experiments, the cells were pretreated with HCQ (10 mM) prior to CIRT.
Clonogenic assay
The cells were irradiated with carbon ion beams at (doses of) 0.5, 1, or 2 Gy. Immediately after irradiation, the cells were detached and counted. Single cells were reseeded into culture dishes at very low densities. A few weeks after culture, the dishes were fixed and stained with 0.5% crystal violet (Sigma-Aldrich, STL, USA). Colonies were counted after staining. Survival fractions were calculated as follows: number of colonies divided by the number of seeded cells and normalized to the plating efficiency of unirradiated control cells (0 Gy). The resulting dataset was subjected to the linear-quadratic regression.
Apoptosis assay
On days 1 or 3 after carbon-ion beam irradiation, apoptotic cells were measured via flow cytometry. We detected the appearance of phosphatidylserine on the plasma membrane during apoptosis by staining with Annexin V-FITC (Thermo Fisher Scientific, MA, USA) [25]. Propidium iodide (PI) (Sigma-Aldrich, STL, USA) does not permeate the plasma membranes of live cells. When the plasma membrane is damaged by changes in its composition, PI enters into the cytoplasm and nucleus. Annexin V and PI staining can distinguish between early apoptotic events (Annexin V+, PI-) and late apoptotic events (necrosis) (Annexin V+, PI+) [25].
Cell cycle assay
On day 3 after carbon-ion beam irradiation, dishes were fixed and treated with RNase A (final 250 mg/mL) and incubated at 37 °C for 15 min. The cells were stained with PI solution (1 mg/mL) at 4 °C for 30 min. DNA in cells binds to PI stoichiometrically. Flow cytometry analysis was conducted to create a histogram of DNA content distribution across each step of the cell cycle.
Western blotting analysis
Cells were harvested, washed with phosphate-buffered saline, and lysed using the tissue protein extraction reagent (Thermo, IL, USA) supplemented with protease inhibitors (Cell Signaling Technology, MA, USA). Protein concentrations were measured using a TaKaRa BCA Protein Assay Kit (TAKARA BIO INC, Japan). Equal amounts of proteins were dissolved in sodium dodecyl sulfate-polyacrylamide gel electrophoresis sample loading buffer and electrophoresed on a polyacrylamide gel (10 or 5–20% gradient gel). After electrophoresis, the proteins were electrotransferred to 0.45-mm polyvinylidene fluoride membranes (ImmobilonR-P Transfer Membrane, Merck Millipore, MA, USA). Immunoblotting was performed using each antibody and detected using the ECL-Select reagent (Amersham, MA, USA).
Mouse experiments
Female C57BL/6J mice (6–8 weeks old) were purchased from Japan SLC, Inc. All animal experiments were performed under specific pathogen-free conditions in accordance with the guidelines of the Institutional Animal Care Committee of the Hyogo College of Medicine (Hyogo, Japan) (protocol number: 20-060).
Pan02 cells were cultured in four groups: no treatment (0 Gy), HCQ alone, CIRT (2 Gy) alone, or HCQ in combination with CIRT 2 Gy. The day after CIRT, cells were harvested and injected with 2.0 x 106 cells per shot to mice on both flanks. Tumor volume was calculated using the following formula: (long diameter) × (short diameter)2/2 [26]. Four weeks after inoculation, mice were euthanized, and their tumors were carefully dissected and weighed.
Patients
Thirty-eight patients with pancreatic cancer did not receive neoadjuvant therapy, while eight patients received neoadjuvant chemotherapy (NAC) (gemcitabine) between 2014 to 2018. Six patients with pancreatic cancer received both NAC (gemcitabine with nab-paclitaxel and/or FOLFIRINOX) [27] and CIRT (total 55.2 Gy by 12 fractionations or total 36.8 Gy by 8 fractionations) between 2019 and 2022. After receiving each treatment, the patients in the three groups underwent pancreaticoduodenectomy or distal pancreatectomy. All research protocols in this study using clinical specimens were approved by the Ethics Committee of Hyogo College of Medicine (approval number: 202208-054).
Immunohistochemistry
Immunohistochemical analysis was performed on pancreatic cancer specimens, as previously described [28]. Briefly, tissue specimens were fixed in 10% Formalin Neutral Buffer Solution (FUJIFILM Wako Chemicals, 062-01661) and embedded in paraffin wax before sectioning. For epitope retrieval, the sections were heated in 10 mM Citrate Buffer (pH 6.0) at 95 °C for 40 min using a water bath. Peroxidase blocking was performed in 0.3% H2O2 / MeOH for 30 min at room temperature. The sections were preincubated with a serum-free protein block (Agilent Technologies X0909) for 30 min at room temperature, and primary antibodies were added to each slide at 4°C overnight. Secondary antibody staining was performed using EnVision +/ HRP, Rabbit (Agilent Technologies K4003) for 30 min at room temperature. HRP activity was visualized using chromogenic substrate 3, 3’-Diaminobenzidine (DAB). Staining was visualized under a Nikon Eclipse Ni-U microscope (NIKON CORPORATION, Tokyo, Japan). The histochemical intensity was analyzed using the ImageJ software version 1.52a (SciJava software ecosystem’s open-source software project) [29, 30].
Statistical analysis
Multiple comparisons among three or four groups were analyzed using Tukey’s test with free web software (www.gen-info.osaka-u.ac.jp/MEPHAS/tukey.html). Statistical significance was set at P < 0.05.
Results
Carbon ion irradiation decreases the survival of human pancreatic cancer cells
To test the cytocidal effects of CIRT on pancreatic cancer cell lines (MIA PaCa-2, BxPC3 and PANC 1), we performed a clonogenic assay. On behalf for the pancreatic cancer cell lines, PANC 1 results were shown (Fig. 1A). The colony number was significantly diminished in CIRT-treated cells compared with that in the control (0 Gy). The surviving fraction decreased in a dose-dependent manner (Fig. 1B). The other two cell lines showed the same tendency (data not shown). These results indicate that carbon-ion irradiation reduced the clonogenicity of pancreatic cancer cells. To investigate whether the CIRT diminution of clonogenicity is associated with apoptotic cell death, we performed an apoptosis assay 72 h after irradiation. The PANC I results for the pancreatic cancer cell lines are shown (Fig. 1C). Apoptotic cells (annexin V+, PI-) and necrotic cells (annexin V+, PI+) were detected by flow cytometry. The apoptotic cell ratio gradually increased in a dose-dependent manner up to 2 Gy, with a sharp increase after 4 Gy irradiation (Fig. 1D). Consistent with the apoptotic cell ratio, the necrotic cell ratio increased in a similar manner (Fig. 1E). Other pancreatic cancer cell lines showed slightly different apoptosis and necrosis ratios, but the trend was the same as that of PANCI (data not shown). These results suggest that carbon ion irradiation has cytocidal activity via the activation of apoptosis, resulting in necrosis. Cell proliferation and apoptosis are known to be highly regulated by cell cycle [31]. To investigate whether CIRT affects the cell cycle, we performed a cell cycle assay in BxPC3 cells 72 h after carbon-ion irradiation. Irradiation increased G2/M peak concentration in a dose-dependent manner (Fig. 1F). The G2/M ratio also increased in a dose-dependent manner (Fig. 1G). These results suggest that carbon-ion irradiation arrested the cell cycle at the G2/M checkpoint.

Fig. 1: Carbon ion irradiation suppress tumorigenicity in pancreatic cancer cells. (A) PANC1 cells were carbon ion irradiated at the indicated dose (LET: 80 KeV/m), and same numbers of cells were seeded into a 6-well plate. Fourteen days after irradiation, colony numbers were counted for calculation of surviving fractions (B). (C) BxPC3 cells were carbon ion irradiated at the indicated dose (LET: 80 KeV/m). Three days after irradiation, apoptotic and necrotic cells were detected via flow cytometry. Apoptotic cell ratio (D) and necrotic cell ratio (E) are shown. (F) BxPC3 cells were carbon ion irradiated at the indicated dose (LET: 80 KeV/m). Three days after irradiation, cell cycle assay was performed. Histogram shows the DNA content distribution across the steps of the cell cycle. G2/M ratio is shown (G). Representative photos and data are shown (A, C, F). Similar results were obtained in two other experiments. Data are presented as the mean ± standard error of the mean (SEM) of triplicate (B, D, E).
HCQ treatment increases the radiosensitivity of human pancreatic cancer cells
To investigate whether autophagy inhibition increased radiosensitivity, we performed an apoptosis assay following CIRT with or without HCQ treatment. On behalf of the pancreatic cancer cell lines, PANC 1 results are shown (Fig. 2A and B). Consistent with the previous data (Fig. 1C–E), CIRT induced apoptosis in a dose-dependent manner (blue bar). Notably, HCQ alone did not induce apoptosis (orange vs. blue bars at 0 Gy). Nonetheless, HCQ pre-treatment further enhanced the apoptosis ratio when combined with CIRT (Fig. 2A and B). Similarly, the necrotic cell ratio was increased by HCQ plus CIRT (orange bar) compared with CIRT alone (blue bar) (Fig. 2C). The other two pancreatic cancer cell lines showed a tendency similar to that of Panc 1 (Fig. S1). Thus, HCQ and CIRT exerted synergistic cytocidal actions against human pancreatic cancer cells. To confirm this, we performed western blot analysis for apoptosis-relevant proteins. Paclitaxel (100 nM) was used as an inducer of apoptosis. HCQ alone did not activate the apoptosis-inducing enzyme caspase-3, which induces its major biological product PARP cleavage (Fig. 2D). However, CIRT alone moderately activated caspase-3 and cleaved PARP (Fig. 2D). Consistent with the results of flow cytometry (Fig. 2A–C), CIRT and HCQ strongly induced caspase 3 activation and PARP cleavage (Fig. 2D). Taken together, these results suggest that CIRT synergizes with HCQ to induce cell death. Therefore, HCQ may serve as a radiosensitizer.
To investigate whether CIRT plus HCQ diminished cell survival, we performed a clonogenic assay using BxPC3 cells (Fig. 2E). The combination of CIRT and HCQ significantly reduced the colony number compared with CIRT alone, and the survival fraction angle of cells treated with 2 Gy CIRT in combination with HCQ (orange line) was significantly steeper than that of cells treated with CIRT alone (blue line) (Fig. 2F). These results suggest that inhibition of autophagy increases the RBE of CIRT.
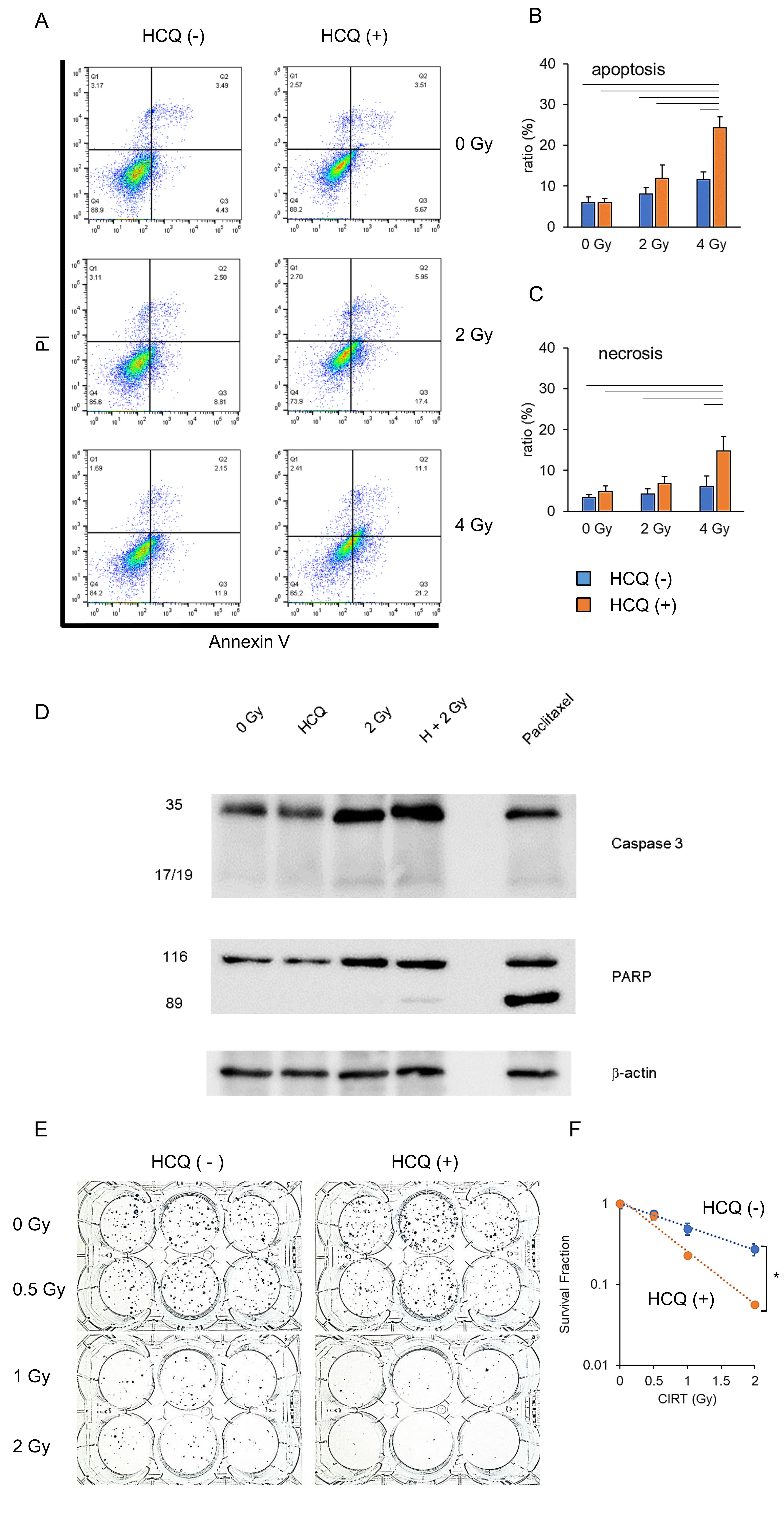
Fig. 2: Hydroxychloroquine (HCQ) treatment enhances the cytotoxicity of carbon ion irradiation for pancreatic cancer. (A) PANC1 cells were carbon ion irradiated at the indicated dose (LET: 80 KeV/m) with or without HCQ (10 M). Three days after irradiation, apoptotic and necrotic cells were detected via flow cytometry. Apoptotic cell ratio (B) and necrotic cell ratio (C) are shown. Blue bar indicates no HCQ treatment. Orange bar indicates HCQ treatment. (D) BxPC3 cells received five kinds of treatments (0 Gy, HCQ only, CIRT 2 Gy only, CIRT 2 Gy in combination with HCQ, and paclitaxel [100 nM]) for 24 h. They were harvested and lysed for western blotting analysis. Caspase-3, cleaved-poly(ADP ribose) polymerase (PARP), and -actin (loading control) were examined. (E) BxPC3 cells were carbon ion irradiated at the indicated dose (LET: 80 KeV/m), and same numbers of cells were seeded into a 6-well plate. Fourteen days after irradiation, colony numbers were counted for calculation of surviving fractions (F). Blue line indicates no HCQ treatment. Orange line indicates HCQ treatment. (A, D, E). Representative photos and data are shown. Similar results were obtained in two other experiments. Data are presented as the mean ± SEM of triplicate (B, C, F).
Carbon ion irradiation simultaneously inhibits the constitutive activation of the RAS-mediated signaling pathway, while activating autophagy
Approximately 90% of pancreatic cancers harbor KRAS mutations. ERK is downstream of KRAS signaling and activated ERK (phosphorylated (p)-ERK) is closely related to cell proliferation. To address other mechanisms underlying the anticancer properties of CIRT, we performed western blot analysis for ERK activation, a signature of mutant KRAS (Fig. 3). Consistent with a previous report [32], MIA PaCa-2 cells showed constitutive ERK phosphorylation under normal conditions. Upon CIRT, the cells displayed a reduction in phosphorylated ERK levels in a dose-dependent manner. However, total ERK expression levels were not affected by CIRT (Fig. 3). These results suggested that CIRT attenuates mutant KRAS-mediated constitutive ERK activation. Inversely, the levels of autophagy detected by LC-3 activation increased after the treatment of pancreatic cancer cells with CIRT in a dose-dependent manner (Fig. 3). Thus, carbon-ion irradiation activates autophagy for cell survival. These results suggest that carbon-ion irradiation produces therapeutically inverse effects on pancreatic cancer cells.

Fig. 3: Carbon ion irradiation attenuates KRAS signaling and activates autophagy in pancreatic cancer cells. MIA PaCa-2 cells were irradiated with carbon ions at the indicated dose (LET: 80 KeV/m). Two hours after irradiation, cells were harvested and lysed for western blotting analysis. Protein levels of extracellular signal-regulated kinase (ERK), p-ERK, LC-3 (autophagy marker), and -actin (loading control) were determined. Representative images are shown. Similar results were obtained in two other experiments.
Carbon ion irradiation together with HCQ attenuates the tumor growth in isograft models
To investigate the effect of CIRT in combination with HCQ on tumor formation in mice, we generated a murine pancreatic cancer isograft model. To estimate the optimal CIRT dose for Pan02 cell death, we performed an apoptosis assay 24 h after CIRT in vitro (Fig. 4A). CIRT alone (blue bar) increased the apoptotic ratio in a dose-dependent manner (Fig. 4B). Pan02 cells showed an additive apoptosis ratio upon HCQ plus CIRT treatment (orange bar). CIRT alone (blue bar) did not increase the necrosis ratio, whereas CIRT in combination with HCQ (orange bar) increased the necrosis ratio in a dose-dependent manner (Fig. 4C). Consistent with human pancreatic cell lines (Fig. 2), 2 Gy CIRT plus HCQ efficiently killed the Pan02 cells. Thus, we generated an isograft model using Pan02 cells treated with 2 Gy CIRT and/or HCQ in vitro . Pan02 cells were prepared after four types of treatments:1 untreated control, 0 Gy; 2 HCQ alone, 3 CIRT 2 Gy alone, and 4 HCQ plus CIRT 2 Gy. Then, 24 h after incubation, the Pan02 cells were inoculated into the syngeneic mice on both franks. The mice were euthanized 28 days after the isograft, and their tumors were carefully dissected, sized, and weighed. As shown in Fig. 4D–F, the tumor size and weight in the HCQ plus CIRT group were minimized among the four groups. The combination treatment attenuated tumor formation compared to the single treatment (HCQ treatment only or CIRT 2 Gy only). These results suggest that CIRT and HCQ protect against tumor formation in vivo .
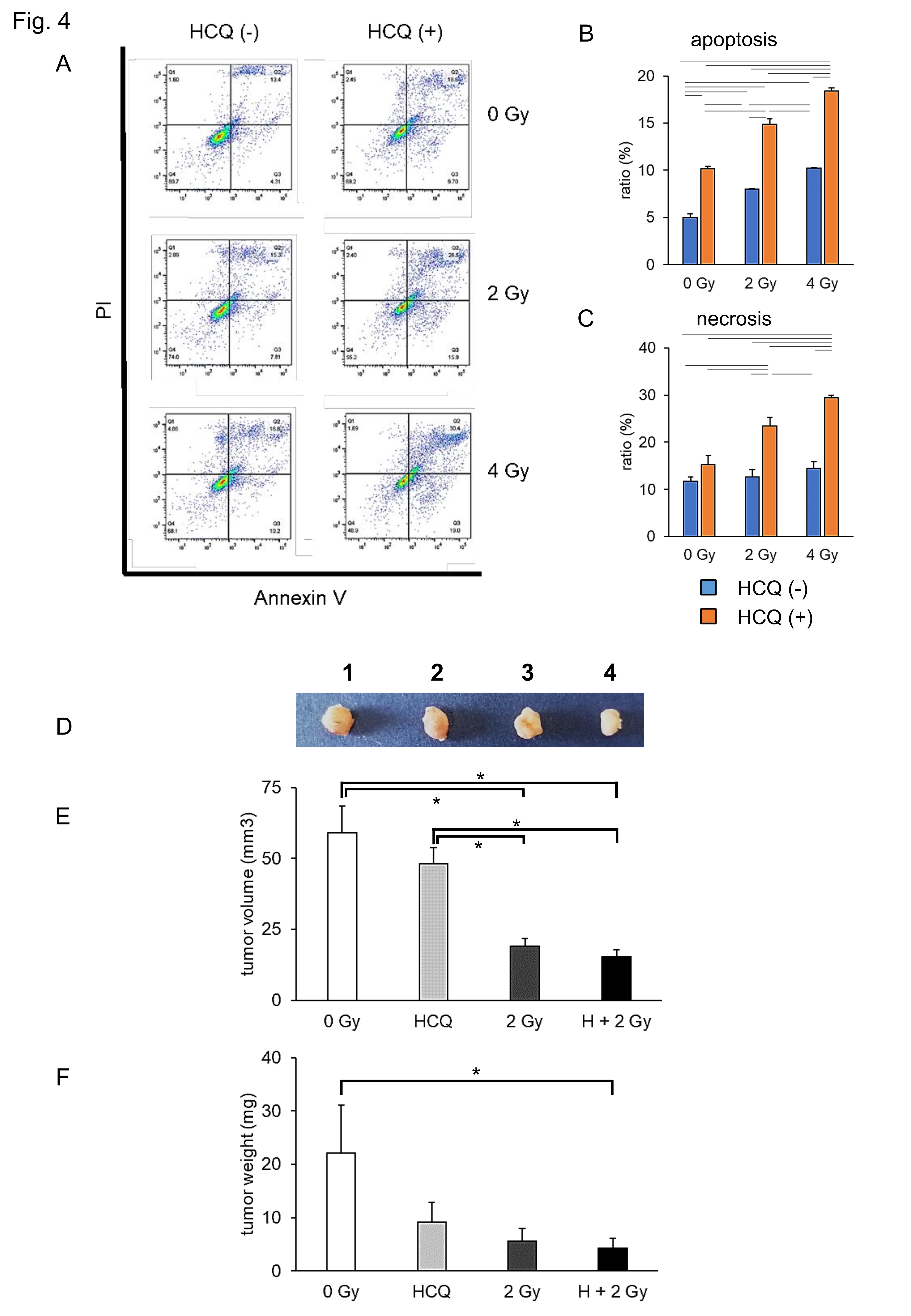
Fig. 4: HCQ treatment enhances the suppression of tumor formation by carbon ion irradiation in isograft models. (A) Pan 02 cells were carbon ion irradiated at the indicated dose (LET: 80 KeV/m) with/without HCQ. Twenty four hours after irradiation, apoptotic and necrotic cells were detected via flow cytometry. Apoptotic cell ratio (B) and necrotic cell ratio (C) are shown. Blue bar indicates no HCQ treatment. Orange bar indicates HCQ treatment. (D) Pan02 cells were prepared after four kinds of treatment: untreated control 0 Gy, HCQ alone, CIRT 2 Gy alone, and HCQ plus CIRT 2 Gy. Twenty four hours after incubation, Pan02 cells were inoculated in the syngeneic mice on both flanks. Twenty eight days after isografting, tumors were isolated (photo), measured (B), and weighted (C). Representative photos and data are shown (A, D). Similar results were obtained in two other experiments. Data are presented as the mean ± SEM of triplicate (B, C, E, F).
Neoadjuvant therapy together with CIRT abolishes autophagy in human pancreatic cancer cells
Thus far, all results indicate that CIRT alone cannot fully exert anticancer action against pancreatic cancer cells by its parallel induction of pro-cancerous autophagy, prompting us to investigate whether this is also the case for patients with pancreatic cancer. Currently, patients with resectable pancreatic cancer receive NAC prior to surgery when the patient is amenable to treatment (from 2019, Clinical Practice Guidelines in Japan) [33]. We measured the activities of autophagy in the surgical samples of patients who received NAC followed by CIRT and compared them with those of patients who received NAC alone. Eight patients received NAC alone, while 6 patients received NAC followed by CIRT before surgery. To investigate whether NAC itself affects the activation of autophagy, we performed the same staining on surgical samples from 38 pancreatic cancer patients without NAC or CIRT (control group). To measure the activation of autophagy, we performed immunohistochemistry for molecules relevant to autophagy: LC3B and p62 (SQSTM1) [34]. Representative images are shown in Fig. 5A. To measure the density of these molecules, we performed image processing using immunohistochemistry images (38 specimens from the control group, eight specimens from the NAC group, and six specimens from NAC plus CIRT; thus, 52 pancreatic cancer specimens in total). Image analyses revealed that there was no statistically significant difference in LC3B or p62 between the NAC and control groups (Fig. 5B and C), indicating that NAC alone did not profoundly affect autophagy activation. In contrast, NAC plus CIRT treatment reduced LC3B and p62 expression levels compared to NAC alone, but not significantly. These results suggest that CIRT alleviates autophagy activity in patients. Thus, in contrast to CIRT in cancer cells, CIRT in patients profoundly protected against the induction of autophagy, presumably leading to the effective elimination of pancreatic cancer. Therefore, neoadjuvant therapy together with CIRT can abolish autophagy in human pancreatic cancer.
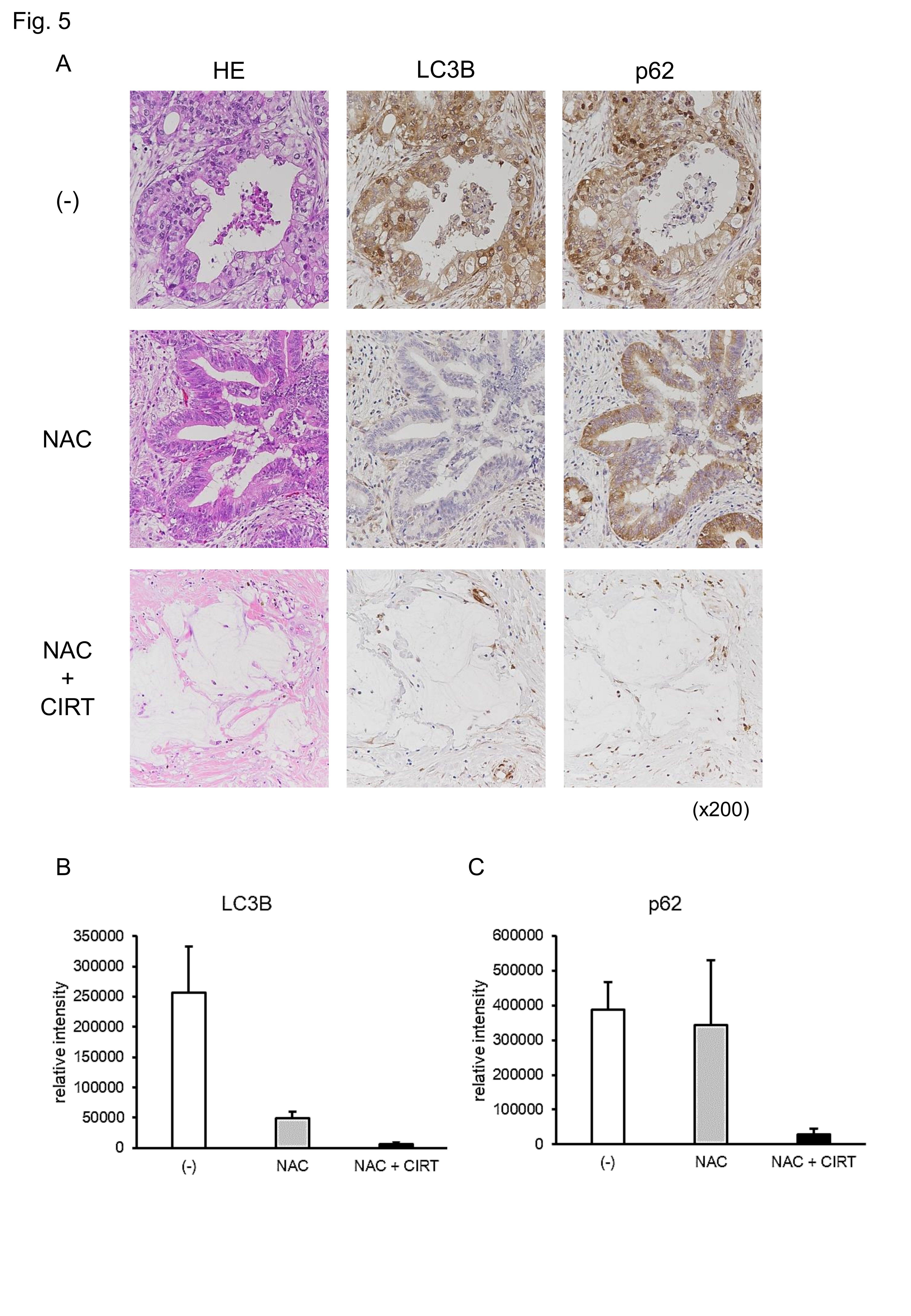
Fig. 5: Neoadjuvant therapy attenuates the expression of autophagy-related molecules. (A) Immunohistochemistry was performed on the surgical specimens of patients with pancreatic cancer without neoadjuvant therapy, with neoadjuvant chemotherapy (NAC), or with NAC plus carbon ion radiotherapy (CIRT) using LC3B and p62 antibodies. (B) LC3B staining intensity was analyzed. (C) p62 staining intensity was analyzed. Control (-), NAC, and NAC plus CIRT groups included, 38, 8, and 6 patients, respectively. Representative photos re shown (A). Data are presented as the mean ± SEM of each group sample (B, C).
Discussion
The present study evaluated the efficacy of CIRT in pancreatic cancer in vitro and in an isograft model. The cytocidal action of CIRT is caused by double-stranded DNA breaks. DNA damage may induce various effects in cancer cells. We observed that cell cycle arrest was induced by CIRT in pancreatic cancer cells (Fig. 1F). Cell cycle progression is highly regulated by cell cycle checkpoints. The G2/M checkpoint is activated by CIRT-induced DNA damage. Cell cycle arrest by CIRT may be associated with the loss of cell proliferation, poor clonogenicity (Fig. 1A and B), and apoptotic cell death, followed by necrosis (Fig. 1C–E).
Autophagy is a lysosome-mediated degradation process [34]. Recently, some studies have reported that activation of autophagy leads to survival when various types of cancer cells are exposed to radiation [18-21]. Radiation induces the bi-directional cell death and cell survival signaling in cancer cells. Therefore, we hypothesized that the inhibition of cell survival induced by autophagy enhances the cytotoxic effects of CIRT in pancreatic cancer. HCQ is an antimalarial drug that is used to treat certain types of autoimmune diseases (lupus, rheumatoid arthritis). Thus, HCQ can be used to inhibit autophagy [35]. We showed that HCQ treatment significantly increased the cell cytotoxicity induced by CIRT (Fig. 2A–C). Similarly, as shown in Fig. 2E and F, the clonogenicity of pancreatic cancer cells was efficiently suppressed by CIRT in combination with HCQ. Interestingly, in Pan02 in vitro apoptosis assay (Fig. 4C), CIRT alone did not increase the necrosis ratio, but CIRT in combination with HCQ increased the necrosis ratio in a dose-dependent manner. It seems that autophagy complemented necrosis when combined with CIRT only. In the Pan02 isograft model, HCQ boosted the tumor suppressor effects of CIRT (Fig. 4D–F). These results suggest that the autophagy inhibitor, HCQ, can be used as a potent radiosensitizer of CIRT. However, HCQ has several limitations that must be addressed before its use in clinical trials. First, HCQ is trapped and accumulated in lysosomes to prevent lysosomal acidification that is necessarily required for the downstream autolysosomal degradation [36], which eventually inhibits many cellular processes, such as proteolytic degradation and post-translational modification of proteins. Second, HCQ is not an autophagy-specific inhibitor and also has anti-inflammatory and immunomodulatory effects [36]. Therefore, we cannot exclude its other pharmacological actions of HCQ. Further research is required to clarify these limitations and facilitate effective clinical evaluation of this therapy.
Reportedly, serum- and glucocorticoid-inducible kinase1 (SGK1) is involved in various pathophysiological processes including tumor biology [37]. SGK1 is highly expressed in several tumors and induce tumor growth, survival, metastasis and so on, suggesting SGK1 as a promising therapeutic target for cancer [38]. Indeed, several studies revealed that the pharmacological inhibition of SGK1 sensitizes the tumor cells to radio therapy [39, 40]. This may implicate that SGK1 inhibitors might enhance anticancer activity of CIRT.
CIRT induces cell death and survival signals in pancreatic cancer cells by three possible mechanisms. First, autophagy mitigates cell damage stress [41]. Cytotoxic cellular damage by CIRT (such as DNA double-strand break and ROS generation) potently induces autophagy. The autophagic response to cellular damage likely facilitates cell survival via the removal of damaged proteins and organelles. Second, most pancreatic cancers (90%) are KRAS-driven. KRAS mutations activate the microphthalmia/transcription factor E transcription program, which regulates autophagy and lysosomal biogenesis. Under stressful conditions, autophagy scavenges intracellular nutrients, damaged proteins, and organelles to recycle central carbon metabolism, which sustains cell survival [42, 43]. Third, the autophagic flux (serine/threonine kinase 11, AMP-activated protein kinase, and Unc-51-like kinase 1), which is responsible for autophagy induction [32], may be controlled, at least in part, by the KRAS pathway. Kinsey et. al. showed a link between mitogen-activated protein kinase-1/2 inhibition and activation of autophagic flux in pancreatic cancer cells [32, 44, 45]. Similarly, we showed a link between the inactivation of ERK and LC-3 (an autophagy marker) expression by CIRT in pancreatic cancer cells (Fig. 3A).
There were significant differences in the outcomes of CIRT induction of autophagy between cellular CIRT and CIRT in patients. Pancreatic cancer cells express higher levels of autophagy upon CIRT (Fig. 3). In contrast, clinical specimens of pancreatic cancer from patients treated with NAC plus CIRT rarely expressed autophagy-related molecules (Fig. 5). There are several possible reasons for this discrepancy. First, after CIRT of the patients with 12 fractions, most cancer cells appeared to undergo cell death. Nuclear staining was negative in these cells (Fig. 5A). Thus, the absence of autophagy in the cancer cells of patients may be attributed to cell death rather than failure of autophagy induction. Second, 12-fractionated irradiation seems to be fatal for cancer cells. Upon a single CIRT at 2 Gy, autophagy was activated in the pancreatic cancer cells, which rescued their survival fraction from 0.056% to 27.3% (Fig. 2E). The patients received 12 cycles of CIRT. If patients were subjected to 2 Gy irradiation per fractionation, 12 cycles of CIRT resulted in only a 0.000017% survival fraction of cancer cells. Third, CIRT-induced cancer cell mortality depends on the irradiation dose of CIRT (Figs. 1 and 2). The single irradiation dose of CIRT in the patients was 4.6 Gy, which is higher than the radiation dose of cellular CIRT (2 Gy). Therefore, with CIRT (55.2 Gy, 12 fractions), the survival fraction of cancer cells may be much smaller than that with CIRT (24 Gy, 12 fractions). Fourth, CIRT enhances the immune response against pancreatic cancer via activation of antigen-presentation [46] and systemic anti-cancer immunity [47]. These events may overcome the induction of CIRT-induced autophagy, which eventually leads to profound cancer cell death in CIRT-irradiated patients.
Conclusion
Our study demonstrated that the autophagy inhibitor, HCQ, enhanced the cytotoxicity of CIRT. Thus, this combination therapy may be an effective treatment strategy for pancreatic cancer. However, these results need to be validated in clinical trials in the future.
Acknowledgements
We would like to thank everyone involved in this research.
Author Contributions
J.F., M.S., H.T., and S. H. conceptualized the study; M.S., K.Y., K.M., N.I., M.A., T.T., M.I., S.S., and T.K. performed experiments; M.S., H.T., S.H., and J.F. analyzed the data; M.S., H.T., S.H., T.K., S.H., E.H., and J.F. wrote the manuscript and all authors reviewed and approved the final version of the manuscript.
Funding
This work was supported by the Japan Society for the Promotion of Scientific Research (JSPS) KAKENHI Grant Number JP21H03011.
Statement of Ethics
Animal experiments conform to internationally accepted standards and have been approved by the Institutional Animal Care Committee, Hyogo Medical University (approval protocol number: 20-060). All research protocols in this study using clinical specimens were approved by the Ethics Committee of Hyogo Medical University (approval number: 202208-054).
Disclosure Statement
The authors declare no conflicts of interest.
References
| 1 | Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F: Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021;71:209-249.
https://doi.org/10.3322/caac.21660 |
| 2 | Mizrahi JD, Surana R, Valle JW, Shroff RT: Pancreatic cancer. Lancet 2020;395:2008-2020.
https://doi.org/10.1016/S0140-6736(20)30974-0 |
| 3 | Liermann J, Shinoto M, Syed M, Debus J, Herfarth K, Naumann P: Carbon ion radiotherapy in pancreatic cancer: A review of clinical data. Radiother Oncol 2020;147:145-150.
https://doi.org/10.1016/j.radonc.2020.05.012 |
| 4 | Huguet F, Andre T, Hammel P, Artru P, Balosso J, Selle F, Deniaud-Alexandre E, Ruszniewski P, Touboul E, Labianca R, de Gramont A, Louvet C: Impact of chemoradiotherapy after disease control with chemotherapy in locally advanced pancreatic adenocarcinoma in GERCOR phase II and III studies. J Clin Oncol 2007;25:326-331.
https://doi.org/10.1200/JCO.2006.07.5663 |
| 5 | Loehrer PJ, Sr., Feng Y, Cardenes H, Wagner L, Brell JM, Cella D, Flynn P, Ramanathan RK, Crane CH, Alberts SR, Benson AB, 3rd: Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an Eastern Cooperative Oncology Group trial. J Clin Oncol 2011;29:4105-4112.
https://doi.org/10.1200/JCO.2011.34.8904 |
| 6 | Hammel P, Huguet F, van Laethem JL, Goldstein D, Glimelius B, Artru P, Borbath I, Bouche O, Shannon J, Andre T, Mineur L, Chibaudel B, Bonnetain F, Louvet C, Group LAPT: Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients With Locally Advanced Pancreatic Cancer Controlled After 4 Months of Gemcitabine With or Without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA 2016;315:1844-1853.
https://doi.org/10.1001/jama.2016.4324 |
| 7 | Usman RM, Razzaq F, Akbar A, Farooqui AA, Iftikhar A, Latif A, Hassan H, Zhao J, Carew JS, Nawrocki ST, Anwer F: Role and mechanism of autophagy-regulating factors in tumorigenesis and drug resistance. Asia Pac J Clin Oncol 2021;17:193-208.
https://doi.org/10.1111/ajco.13449 |
| 8 | Munoz-Maldonado C, Zimmer Y, Medova M: A Comparative Analysis of Individual RAS Mutations in Cancer Biology. Front Oncol 2019;9:1088.
https://doi.org/10.3389/fonc.2019.01088 |
| 9 | Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, Garcia-Echeverria C, Weissleder R, Mahmood U, Cantley LC, Wong KK: Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008;14:1351-1356.
https://doi.org/10.1038/nm.1890 |
| 10 | Alagesan B, Contino G, Guimaraes AR, Corcoran RB, Deshpande V, Wojtkiewicz GR, Hezel AF, Wong KK, Loda M, Weissleder R, Benes CH, Engelman J, Bardeesy N: Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clin Cancer Res 2015;21:396-404.
https://doi.org/10.1158/1078-0432.CCR-14-1591 |
| 11 | Buglewicz DJ, Banks AB, Hirakawa H, Fujimori A, Kato TA: Monoenergetic 290 MeV/n carbon-ion beam biological lethal dose distribution surrounding the Bragg peak. Sci Rep 2019;9:6157.
https://doi.org/10.1038/s41598-019-42600-4 |
| 12 | Amornwichet N, Oike T, Shibata A, Ogiwara H, Tsuchiya N, Yamauchi M, Saitoh Y, Sekine R, Isono M, Yoshida Y, Ohno T, Kohno T, Nakano T: Carbon-ion beam irradiation kills X-ray-resistant p53-null cancer cells by inducing mitotic catastrophe. PLoS One 2014;9:e115121.
https://doi.org/10.1371/journal.pone.0115121 |
| 13 | Shinoto M, Yamada S, Terashima K, Yasuda S, Shioyama Y, Honda H, Kamada T, Tsujii H, Saisho H, Working Group for Pancreas C: Carbon Ion Radiation Therapy With Concurrent Gemcitabine for Patients With Locally Advanced Pancreatic Cancer. Int J Radiat Oncol Biol Phys 2016;95:498-504.
|
| 14 | Vitolo V, Cobianchi L, Brugnatelli S, Barcellini A, Peloso A, Facoetti A, Vanoli A, Delfanti S, Preda L, Molinelli S, Klersy C, Fossati P, Orecchia R, Valvo F: Preoperative chemotherapy and carbon ions therapy for treatment of resectable and borderline resectable pancreatic adenocarcinoma: a prospective, phase II, multicentre, single-arm study. BMC Cancer 2019;19:922.
https://doi.org/10.1186/s12885-019-6108-0 |
| 15 | Mikkelsen RB, Wardman P: Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 2003;22:5734-5754.
https://doi.org/10.1038/sj.onc.1206663 |
| 16 | Scriven P, Brown NJ, Pockley AG, Wyld L: The unfolded protein response and cancer: a brighter future unfolding? J Mol Med (Berl) 2007;85:331-341.
https://doi.org/10.1007/s00109-006-0150-5 |
| 17 | Ghorai A, Sarma A, Bhattacharyya NP, Ghosh U: Carbon ion beam triggers both caspase-dependent and caspase-independent pathway of apoptosis in HeLa and status of PARP-1 controls intensity of apoptosis. Apoptosis 2015;20:562-580.
https://doi.org/10.1007/s10495-015-1107-3 |
| 18 | Wang F, Tang J, Li P, Si S, Yu H, Yang X, Tao J, Lv Q, Gu M, Yang H, Wang Z: Chloroquine Enhances the Radiosensitivity of Bladder Cancer Cells by Inhibiting Autophagy and Activating Apoptosis. Cell Physiol Biochem 2018;45:54-66.
https://doi.org/10.1159/000486222 |
| 19 | Chen Y, Li X, Guo L, Wu X, He C, Zhang S, Xiao Y, Yang Y, Hao D: Combining radiation with autophagy inhibition enhances suppression of tumor growth and angiogenesis in esophageal cancer. Mol Med Rep 2015;12:1645-1652.
https://doi.org/10.3892/mmr.2015.3623 |
| 20 | Hu JL, He GY, Lan XL, Zeng ZC, Guan J, Ding Y, Qian XL, Liao WT, Ding YQ, Liang L: Inhibition of ATG12-mediated autophagy by miR-214 enhances radiosensitivity in colorectal cancer. Oncogenesis 2018;7:16.
https://doi.org/10.1038/s41389-018-0028-8 |
| 21 | Sailaja GS, Bhoopathi P, Gorantla B, Chetty C, Gogineni VR, Velpula KK, Gondi CS, Rao JS: The secreted protein acidic and rich in cysteine (SPARC) induces endoplasmic reticulum stress leading to autophagy-mediated apoptosis in neuroblastoma. Int J Oncol 2013;42:188-196.
https://doi.org/10.3892/ijo.2012.1678 |
| 22 | Chaurasia M, Gupta S, Das A, Dwarakanath BS, Simonsen A, Sharma K: Radiation induces EIF2AK3/PERK and ERN1/IRE1 mediated pro-survival autophagy. Autophagy 2019;15:1391-1406.
https://doi.org/10.1080/15548627.2019.1582973 |
| 23 | Deer EL, Gonzalez-Hernandez J, Coursen JD, Shea JE, Ngatia J, Scaife CL, Firpo MA, Mulvihill SJ: Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010;39:425-435.
https://doi.org/10.1097/MPA.0b013e3181c15963 |
| 24 | Corbett TH, Roberts BJ, Leopold WR, Peckham JC, Wilkoff LJ, Griswold DP, Jr., Schabel FM, Jr.: Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res 1984;44:717-726.
|
| 25 | Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C: A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods 1995;184:39-51.
https://doi.org/10.1016/0022-1759(95)00072-I |
| 26 | Faustino-Rocha A, Oliveira PA, Pinho-Oliveira J, Teixeira-Guedes C, Soares-Maia R, da Costa RG, Colaco B, Pires MJ, Colaco J, Ferreira R, Ginja M: Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab Anim (NY) 2013;42:217-224.
https://doi.org/10.1038/laban.254 |
| 27 | Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, Bennouna J, Bachet JB, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M, Groupe Tumeurs Digestives of U, Intergroup P: FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817-1825.
https://doi.org/10.1056/NEJMoa1011923 |
| 28 | Itou RA, Uyama N, Hirota S, Kawada N, Wu S, Miyashita S, Nakamura I, Suzumura K, Sueoka H, Okada T, Hatano E, Tsutsui H, Fujimoto J: Immunohistochemical characterization of cancer-associated fibroblasts at the primary sites and in the metastatic lymph nodes of human intrahepatic cholangiocarcinoma. Hum Pathol 2019;83:77-89.
https://doi.org/10.1016/j.humpath.2018.08.016 |
| 29 | Sudo M, Iida K, Tsutsui H, Mitani K, Jimbo M, Hatano E, Fujimoto J: Blockade of Tumor Necrosis Factor by Etanercept Prevents Postoperative Adhesion Formation in Mice. Cell Physiol Biochem 2020;54:1041-1053.
https://doi.org/10.33594/000000286 |
| 30 | Sudo M, Xu J, Mitani K, Jimbo M, Tsutsui H, Hatano E, Fujimoto J: Antithrombin Together with NETs Inhibitor Protected Against Postoperative Adhesion Formation in Mice. Cell Physiol Biochem 2021;55:400-412.
https://doi.org/10.33594/000000392 |
| 31 | Vermeulen K, Berneman ZN, Van Bockstaele DR: Cell cycle and apoptosis. Cell Prolif 2003;36:165-175.
https://doi.org/10.1046/j.1365-2184.2003.00267.x |
| 32 | Huart C, Chen JW, Le Calve B, Michiels C, Wera AC: Could Protons and Carbon Ions Be the Silver Bullets Against Pancreatic Cancer? Int J Mol Sci 2020;21
https://doi.org/10.3390/ijms21134767 |
| 33 | Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT, Yap JT, Burrell LD, Lum DH, Whisenant JR, Gilcrease GW, 3rd, Cavalieri CC, Rehbein KM, Cutler SL, Affolter KE, Welm AL, Welm BE, Scaife CL, Snyder EL, McMahon M: Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med 2019;25:620-627.
https://doi.org/10.1038/s41591-019-0367-9 |
| 34 | Motoi F, Kosuge T, Ueno H, Yamaue H, Satoi S, Sho M, Honda G, Matsumoto I, Wada K, Furuse J, Matsuyama Y, Unno M, Study Group of Preoperative Therapy for Pancreatic C, Japanese Study Group of Adjuvant Therapy for Pancreatic c: Randomized phase II/III trial of neoadjuvant chemotherapy with gemcitabine and S-1 versus upfront surgery for resectable pancreatic cancer (Prep-02/JSAP05). Jpn J Clin Oncol 2019;49:190-194.
https://doi.org/10.1093/jjco/hyy190 |
| 35 | Mathew R, Karantza-Wadsworth V, White E: Role of autophagy in cancer. Nat Rev Cancer 2007;7:961-967.
https://doi.org/10.1038/nrc2254 |
| 36 | Al-Bari MAA: Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol Res Perspect 2017;5:e00293.
https://doi.org/10.1002/prp2.293 |
| 37 | Leong ML, Maiyar AC, Kim B, O'Keeffe BA, Firestone GL: Expression of the serum- and glucocorticoid-inducible protein kinase, Sgk, is a cell survival response to multiple types of environmental stress stimuli in mammary epithelial cells. J Biol Chem 2003;278:5871-5882.
https://doi.org/10.1074/jbc.M211649200 |
| 38 | Zhu R, Yang G, Cao Z, Shen K, Zheng L, Xiao J, You L, Zhang T: The prospect of serum and glucocorticoid-inducible kinase 1 (SGK1) in cancer therapy: a rising star. Ther Adv Med Oncol 2020;12:1758835920940946.
https://doi.org/10.1177/1758835920940946 |
| 39 | Towhid ST, Liu GL, Ackermann TF, Beier N, Scholz W, Fuchss T, Toulany M, Rodemann HP, Lang F: Inhibition of colonic tumor growth by the selective SGK inhibitor EMD638683. Cell Physiol Biochem 2013;32:838-848.
https://doi.org/10.1159/000354486 |
| 40 | Liu G, Honisch S, Liu G, Schmidt S, Pantelakos S, Alkahtani S, Toulany M, Lang F, Stournaras C: Inhibition of SGK1 enhances mAR-induced apoptosis in MCF-7 breast cancer cells. Cancer Biol Ther 2015;16:52-59.
https://doi.org/10.4161/15384047.2014.986982 |
| 41 | Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, Mautner J, Tonon G, Haigis M, Shirihai OS, Doglioni C, Bardeesy N, Kimmelman AC: Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011;25:717-729.
https://doi.org/10.1101/gad.2016111 |
| 42 | Galluzzi L, Bravo-San Pedro JM, Demaria S, Formenti SC, Kroemer G: Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol 2017;14:247-258.
https://doi.org/10.1038/nrclinonc.2016.183 |
| 43 | Lee CS, Lee LC, Yuan TL, Chakka S, Fellmann C, Lowe SW, Caplen NJ, McCormick F, Luo J: MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc Natl Acad Sci U S A 2019;116:4508-4517.
https://doi.org/10.1073/pnas.1817494116 |
| 44 | White E: Blockade of RAF and autophagy is the one-two punch to take out Ras. Proc Natl Acad Sci U S A 2019;116:3965-3967.
https://doi.org/10.1073/pnas.1900800116 |
| 45 | Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD, Tomar G, Papke B, Hobbs GA, Yan L, Hayes TK, Diehl JN, Goode GD, Chaika NV, Wang Y, Zhang GF, Witkiewicz AK, Knudsen ES, Petricoin EF, 3rd, Singh PK, Macdonald JM, Tran NL, Lyssiotis CA, Ying H, Kimmelman AC, Cox AD, Der CJ: Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med 2019;25:628-640.
https://doi.org/10.1038/s41591-019-0368-8 |
| 46 | Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, Sohn ASW, Mukhopadhyay S, Lin EY, Parker SJ, Banh RS, Paulo JA, Wen KW, Debnath J, Kim GE, Mancias JD, Fearon DT, Perera RM, Kimmelman AC: Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020;581:100-105.
https://doi.org/10.1038/s41586-020-2229-5 |
| 47 | Takahashi Y, Yasui T, Minami K, Tamari K, Hayashi K, Otani K, Seo Y, Isohashi F, Koizumi M, Ogawa K: Carbon ion irradiation enhances the antitumor efficacy of dual immune checkpoint blockade therapy both for local and distant sites in murine osteosarcoma. Oncotarget 2019;10:633-646.
https://doi.org/10.18632/oncotarget.26551 |