Dysregulation of Aquaporin-3 and Glyceryl Glucoside Restoring Action in Hidradenitis Suppurativa in Vitro Models
bDepartment of Neuroscience, Scuola Internazionale Superiore di Studi Avanzati (SISSA), Trieste, Italy,
cINSERM, IMRB, Translational Neuropsychiatry, University Paris Est Créteil, Créteil, France,
dDepartment of Advanced Diagnostics, Institute for Maternal and Child Health - IRCCS Burlo Garofolo, Trieste, Italy,
eDepartment of Pharmacy, University of Salerno, Salerno, Italy,
fDermatology Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy,
gDepartment of Pathophysiology and Transplantation, Università degli Studi di Milano, Milan, Italy,
hMaternal-Neonatal Department, Institute for Maternal and Child Health - IRCCS Burlo Garofolo, Trieste, Italy,
iDepartment of Biosciences, Biotechnologies and Environment, University of Bari Aldo Moro, Bari, Italy,
jLaboratory of Animal Research Center (LARC), Qatar University,
kDepartment of Pediatrics, Institute for Maternal and Child Health - IRCCS Burlo Garofolo, Trieste, Italy
Keywords
Abstract
Background/Aims:
Aquaporin-3 (AQP3) is an aquaglyceroporin and peroxiporin that plays a crucial role in skin barrier homeostasis. Dysregulated AQP3 expression has been observed in different inflammatory skin conditions. Hidradenitis Suppurativa (HS) is an autoinflammatory keratinization disease that typically appears between 10 and 21 years of age, characterized by alteration of skin barrier homeostasis.Methods:
To evaluate in vitro the role of AQP3 in the development of HS, we performed real-time PCR and Western blot to analyze gene and protein levels in human keratinocyte cell lines knock-out (KO) for NCSTN and PSENEN genes, simulating genetic-associated HS. Additionally, we investigated the impact of Glyceryl Glucoside (GG) on biological processes by performing MTT, scratch, proliferation assays and proteome studies.Results:
We detected a significant decrease of the levels of AQP3 gene and protein in KO cell lines. GG effectively elevated the levels of mRNA and protein, significantly decreased the hyperproliferation rate, and enhanced cell migration in our in vitro model of genetic Hidradenitis Suppurativa. Pathway enrichment analysis further confirmed GG’s role in the migration and proliferation pathways of keratinocytes.Conclusion:
Our results suggest that AQP3 may act as a new novel actor in HS etio-pathogenesis, and GG could be further explored as potential treatment option for managing HS in patients.Introduction
Aquaporin-3 (AQP3) is a membrane channel protein of broad selectivity that facilitates the bidirectional movement of water and a series of small neutral solutes including glycerol, ammonia, urea and hydrogen peroxide [1]. AQP3 is present in different organs and tissues including skin, which is one of the predominant sites of AQP3 expression as high levels are found in the keratinocytes of the stratum basale, spinosum and granulosum. In addition to the important roles exerted in hydric balance and energy homeostasis AQP3 is emerging as a key player in inflammation, redox‐associated signaling, and in different immune processes [2] triggering strong interest for its potential pharmacological relevance as drug target [3]. In the skin, AQP3 is pivotal in skin barrier permeability and skin hydration and seems to have a role in keratinocyte migration, proliferation and epithelial wound healing [4, 5].
Given the crucial role of AQP3 in skin hydration and in various keratinocytes’ processes, aberrations in its expression and location have been found in different inflammatory skin diseases [6]. Specifically, AQP3 dysregulations have been mainly observed in atopic dermatitis, rosacea and psoriasis, leading to different phenotypic outcomes [7–9].
Hidradenitis Suppurativa (HS) is an autoinflammatory keratinization disease, clinically characterized by recurrent painful lesions; it typically appears between 10 and 21 years of age and disproportionately affects women, with a sex ratio of about 1:3 [10–12].
The etiology of HS is multifactorial, involving a close interaction between genetic and environmental factors, as well as immune dysregulation, hormonal alterations and variations in the skin microbiome [13]. Despite recent advances, how these factors shape the pathogenesis of the disease is partially known, although the primum movens seems to be the hyperplasia of the infundibular epithelium, that causes follicular clogging and subsequent stagnation of follicular content, resident bacteria’s propagation and dilatation of the pilosebaceous unit [13], hence triggering the development of an inflammatory environment.
It is known that about one-third of HS patients present a positive family history of the disease, with most loss of function mutations occurring in genes encoding for the γ-secretase complex subunits, particularly presenilin enhancer 2 (PSENEN) and nicastrin (NCSTN). This protease complex mediates intramembranous cleavage of various type I membrane proteins, including Notch receptors [13–15].
HS adversely affects patients’ quality of life through both its clinical skin manifestations and significant negative psychological impacts [16]. Despite the availability of biological drugs to treat patients with Hidradenitis Suppurativa (HS), these treatments yield varying degrees of success and outcomes [15, 17].
All this considered, in order to unravel the possible involvement of AQP3 in the pathogenic scenario of HS, we evaluated its gene expression and protein levels in a HS genetic in vitro model obtained using human keratinocyte cell line (HaCaT) knock-out (KO) for NCSTN and PSENEN genes (NCSTN -/- and PSENEN -/-).
Given the dysregulation observed in our in vitro model of Hidradenitis Suppurativa (HS), we opted to explore the effects of Glyceryl Glucoside (GG), a compound reported to stimulate the transcriptional expression of AQP3 [18]. Our approach was twofold: firstly, to confirm the role of GG in boosting the levels of AQP3, and secondly, to evaluate its broader impact on wound healing and cellular behaviours associated with skin repair. To verify GG’s efficacy in enhancing AQP3 expression, we treated our HS model with GG and measured the subsequent changes in AQP3 levels. This involved precise quantification techniques to ensure accurate assessment of transcriptional activity. The increased expression of AQP3, a protein known for its role in skin hydration and function, indicated GG’s potential as a therapeutic agent for managing HS. In addition to measuring AQP3 levels, we conducted a functional analysis to determine GG’s effectiveness in promoting wound healing. This was done by creating a scratch in a monolayer of cultured keratinocytes, which served as a simulation of a skin lesion; the scratch assay is a well-established method for studying cell migration and wound closure. By monitoring the rate of scratch closure, we were able to gauge the healing capacity of keratinocytes in the presence of GG. Furthermore, we examined the rate of cell proliferation. Finally, to gain deeper insights into the underlying mechanisms, we identified the main signalling pathways involved in these processes; this included the analysis of pathways related to cell migration, proliferation, and survival, which are essential for effective wound repair. By elucidating these pathways, we aimed to uncover the molecular basis of GG’s action and its potential therapeutic benefits for HS patients.
Materials and Methods
Cell lines
HaCaT cells, an immortalized human keratinocyte line, were cultured in low-calcium medium (DMEM, Euroclone , supplemented with 10% of fetal bovine serum, FBS, Euroclone , 0.03 mM CaCl2, Sigma-Aldrich ).
From previous experiments conducted in our laboratory, we obtained knock-out HaCaT cell lines for two different genes, NCSTN (NCSTN -/-) and PSENEN (PSENEN -/-), as described in our previous work [19]. The cells were treated with complete medium supplemented with 4% v/v of Glycoin® natural, a 100% natural sugar derivate Glyceryl Glucoside (GG) by Bitop (https://www.bitop.de/en/products/cosmetic-active-ingredients/glycoin) , who kindly offered us the product.
RNA extraction and Real-time PCR
Gene expression was evaluated with real-time PCR. Briefly, HaCaT cells were cultured at 1.5x105/well in 6-well plates. After 24 hours, medium was changed to complete medium supplemented with or without 4% GG, and following an additional 24 hours cells were lysed with Trizol reagent (TriFast, Euroclone ) to extract RNA. RNA was retrotranscribed with High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems™ ). Real-time PCR was performed with TaqMan probes from Applied Biosystems™ , using CFX Opus 96 Real-Time PCR System (Bio-Rad ).
Western blot
Protein expression was evaluated by western blot. HaCaT cells were cultured at 1.5x105/well in 6-well plates. After 24 hours, the medium was changed with complete medium enriched with or without 4% GG, and after additional 24 hours cells were lysed with RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific ).
Gel electrophoresis was performed using NuPAGE™ 4 to 12%, Bis-Tris in NuPAGE MES SDS Running Buffer (Invitrogen™ ). Proteins were transferred on nitrocellulose membranes (Bio-Rad ) employing the TransBlot® Turbo™ Transfer System (Bio-Rad ) and probed with primary antibodies including anti-AQP3 (Abcam ), anti-HSP90 (Santa Cruz Biotechnology ) and were then incubated with HRP-conjugated secondary antibodies (Cell Signaling ). ClarityTM substrate (Bio-Rad ) was used and chemiluminescent detection was accomplished with the ChemiDoc Imaging System (Bio-Rad ).
MTT assay
Cells were plated at a density of 1x104 cells/well in 96 well plates for the 24 hours testing, while they were cultured at 5x103 cells/well in 96 well plates for the 72 hours time point. After 24 hours, medium was discarded and complete medium supplemented with different concentrations of GG (3%, 4%, 5%, 6%).
Cell viability was performed using the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay (Invitrogen ). The absorbance was measured at 560 nm with GloMax®-Multi Detection System (Promega ).
Scratch assay
Cells were cultured in 96-well microplates at a confluence of 3x104 cells/well 24 hours after seeding, scratch wounds were created mechanically with a pipette tip. Next, media was replaced to one with or without GG at 4%. After another 24 hours, medium was changed to medium without GG, and the assay was carried on for another 48 hours. Plates were analysed with BioTek BioSpa Live Cell Analysis System (Agilent ).
Scratch assay results were indicated as migration rate values, determined using the following equation:
migration rate (mm2/h) = (Premigration area [mm2] — migration area [mm2] / time [hour])
Proliferation assay
To estimate differences in cell proliferation rates after GG treatment we used the xCELLigence Real-Time Cell Analysis (Agilent ) system, due to which cell proliferation was monitored in real time every hour for a total of 72 hours. 3.9x103 cells/well were plated in 96-well microplates and the tracking of proliferation started immediately. After 24 hours, media was changed with media supplemented with or without 4%GG. After another 24 hours, medium was changed to medium without GG, and the assay was carried on for another 48 hours.
For every cell line, the means of every time point of the treated cells were normalized to the mean of the non-treated cells (NT) at the same time point; the result is a fold change of the GG-treated cells relative to the NT.
Proteome
Cell lines have been lysed with the kit EasyPep™ MS Sample Prep Kits (Thermo Fisher Scientific ). After lysis, the proteins were quantified with Bradford assay (Biorad ), following the manufacturers’ protocol, and 80 µg of proteins were digested with the same kit. After digestion and clean-up, mass spectrometry analysis was performed by nanoflow ultra-high performance liquid chromatography-high resolution mass spectrometry using an Ultimate 3000 nanoLC (Thermo Fisher Scientific ) coupled to an Orbitrap Fusion™ Lumos™ (Thermo Fisher Scientific ) with an Easy nano electrospray ion source (Thermo Fisher Scientific ). High resolution mass spectrometry analysis (HRMS) analysis was performed in data dependent acquisition (DDA), with MS1 range 375–1.500 m/z. For data processing, raw MS data were analyzed using Proteome Discoverer v 2.5 (Thermo Fisher Scientific ).
Abundance data normalization, imputation, and differentially expressed proteins (DEP) analysis was carried out using dep R package [20]. Furthermore, the set Differentially expressed proteins was submitted to enrichment analysis, using reactomePA package [21].
Data analysis and statistical tests
Every assay was repeated 3 times, every time in duplicate. When comparing treated to non-treated cells for every cell line, data analyses were performed with non-parametric Mann-Whitney test. Regarding the comparison between different cell lines, different GG concentrations in the MTT assay, and different time points in the proliferation assay, One-way ANOVA followed by Kruskal-Wallis multiple comparison post hoc test was performed.
Results
Cytotoxicity of GG treatments
Different concentrations of GG were evaluated for cytotoxicity to determine the highest concentration that is safe and does not produce adverse effects on the cell lines used. According to MTT assay, the highest concentration of GG bearing non-significant changes in cell vitality in each of the three cell lines, both after 24 (Fig. 1A) and 72 hours (Fig. 1B), is 4% (% cell viability to NT GG 4% 24h, WT: 101.50±8.74; NCSTN -/-: 107.5±4.123; PSENEN -/-: 89.25±5.62. % cell viability to NT GG 4% 72h, WT: 96.5±7.85; NCSTN -/-: 96.8±6.13; PSENEN -/-:91.8±4.50).
Based on these results, all experimental procedures were performed by treating cells with medium supplemented with 4% of GG (herein indicated as GG).
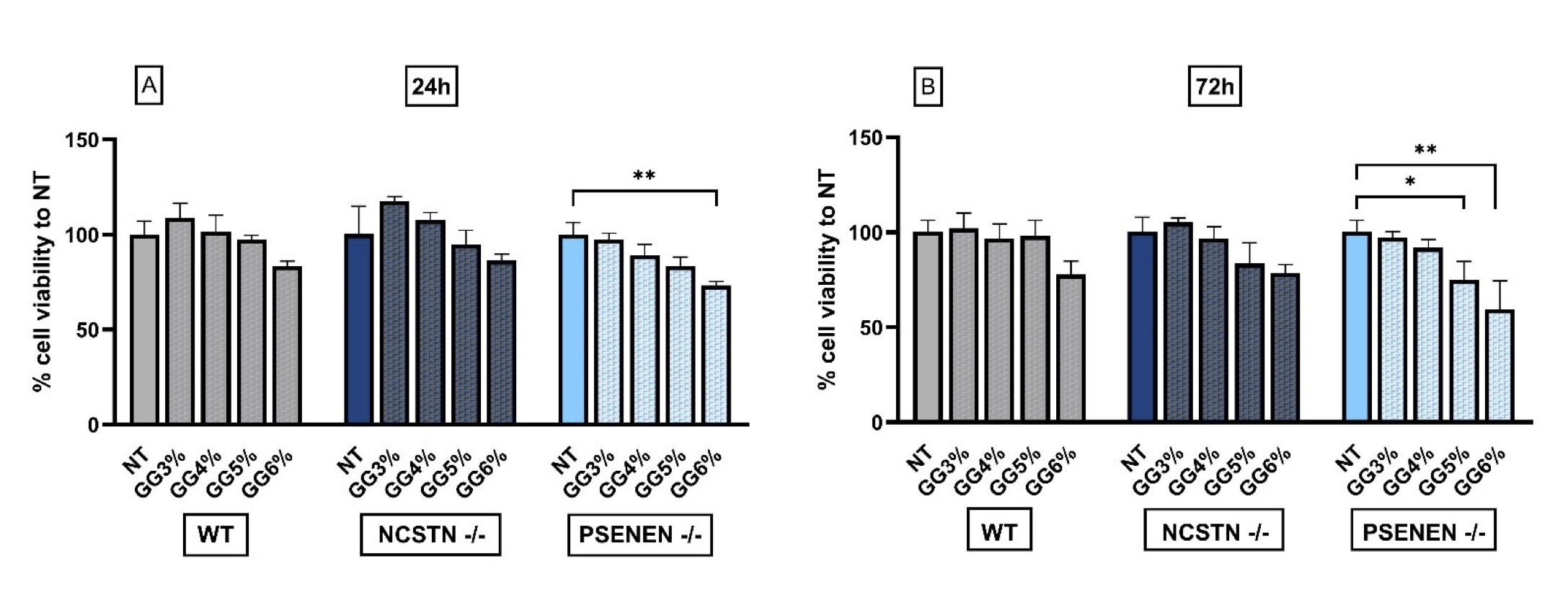
Fig. 1: MTT cytotoxicity assay to evaluate glycerol glucoside effect on HaCaT wild-type, NCSTN -/-, and PSENEN -/-, indicating 4 % GG as the higher concentration without significant toxic effect. (A): % cell viability was evaluated 24 hours after GG treatment by MTT assay. Data are shown as the percentage of levels compared with non-treated cells (NT), considered as 100% of survival. P**=0.0032. (B): % cell viability was evaluated 72 hours after GG treatment by MTT assay. Data are shown as the percentage of levels compared with non-treated cells (NT), considered as 100% of survival. P*=0.0479, P**=0.0049.
Significant decrease of AQP3 gene and protein expression in in vitro HS model
AQP3 gene expression was evaluated by real-time PCR. We detected a significant decrease in AQP3 gene expression in Knock-Out (KO) HaCaT cell lines, NCSTN -/- and PSENEN -/- cells, when compared to wild-type (WT) HaCaT (Fold increase AQP3 mRNA: WT: 0.86±0.25; NCSTN -/- : 0.08±0.07, P<0, 0001; PSENEN -/-: 0.24±0.20; P=0, 0007) (Fig. 2A).
To evaluate whether mRNA decreased expression was correlated with a diminished protein expression we performed Western blot analysis on the same cell lines. We confirmed a significant AQP3 decrease in NCSTN -/- HaCaT cell line when compared to WT cell line, while PSENEN -/- decrease was observed but the result was not statistically significant (Fold Increase AQP3 protein: WT: 1.07±0.22; NCSTN -/- : 0.10±0.02, P=0.0002; PSENEN -/-: 0.35±0.21) (Fig. 2B).
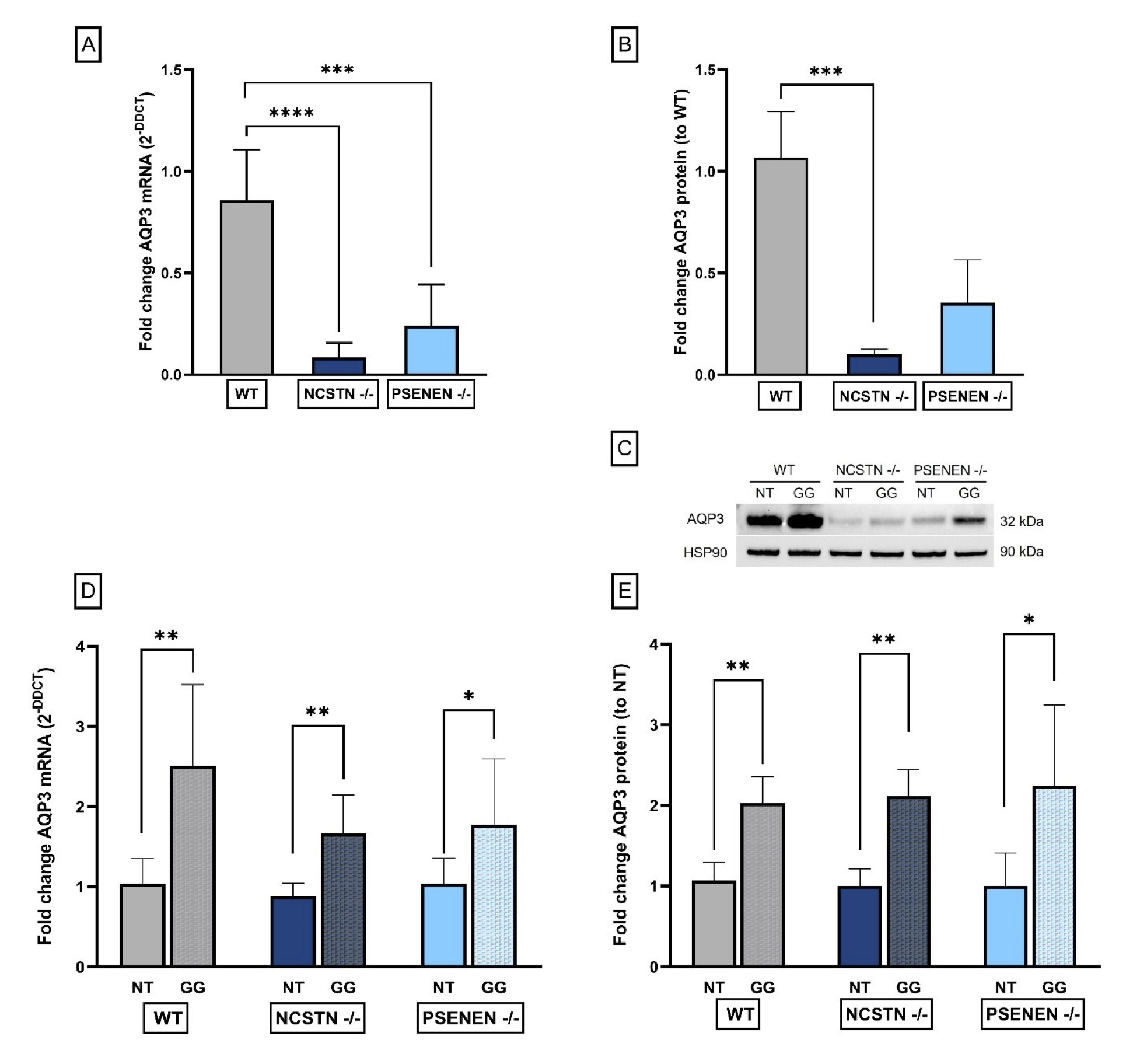
Fig. 2: AQP3 expression levels. (A): Real-time qPCR experiment results expressed as fold increase of AQP3 mRNA levels, reported as DDCt average±SD. P****=0.0001, P***=0.0007. (B): Quantification of AQP3, normalized to HSP90 optical density, reported as a fold change in AQP3 protein levels. P***=0.0002. (C): Western blot showing the levels of AQP3 and HSP90, the latter employed as the housekeeping reference protein. (D): Real-time PCR results expressed as fold increase of AQP3 mRNA levels, reported as DDCt average±SD. P**=0.0043, P**=0.0043, P*=0.0411. (E): Quantification of AQP3, normalized to HSP90 optical density, reported as a fold change in AQP3 protein levels compared to non-treated cells. P**=0.0022, P**=0.0022, P*=0.026.
GG treatment induced an increase in AQP3 gene and protein expression
To evaluate the effect of GG treatment on AQP3 gene and protein expression, we performed real-time qPCR and Western blot experiments on the cell lines, comparing non-treated with treated cells. Following GG treatment, we observed a significant increase in AQP3 gene expression in all tested cell lines, when compared to non-treated conditions (Fold Increase AQP3 mRNA: WT NT: 1.04±0.31; WT GG: 2.51±1.02, P=0.0043; NCSTN -/- NT: 0.88±0.16; NCSTN -/- GG: 1.66±0.48, P=0.0043; PSENEN -/- NT: 1.04±0.32; PSENEN -/- GG: 1.77±0.82, P=0.0411) (Fig. 2D).
We confirmed the same significant trend in AQP3 protein levels after GG treatment in all cell lines compared to non-treated conditions (Fold change AQP3 protein: WT NT: 1.10±0.22; WT GG: 2.00±0.33, P=0.0022; NCSTN -/- NT: 1.00±0.21; NCSTN -/- GG: 2.1±0.34, P=0.0022; PSENEN -/- NT: 1.0±0.41; PSENEN -/- GG: 2.2±1.0, P=0.0260) (Fig. 2E).
GG induced a significant increase in migration rate
As previously established in our earlier research, the healing process for HS lesions is characteristically prolonged [19]. In light of this, a scratch assay was conducted to assess if GG treatment could enhance wound closure rates in HS using an in vitro model of the disease.
As shown in Fig. 3A and 3B, GG was able to significantly increase the migration rate in KO cell lines. Specifically, differences between non-treated and treated cells become significant after 48 and 60 hours from the performance of the mechanical scratch in NCSTN -/- cell line, and after 48 and 72 h in PSENEN -/- cell line (Migration rate: NCSTN -/- NT 48 h: 0.0053±0.00074; NCSTN -/- GG 48 h: 0.0067±0.0010, P=0.0002; NCSTN -/- NT 60 h: 0.0046±0.00052; NCSTN -/- GG 60 h: 0.0057±0.00054, P=0.0070; PSENEN -/- NT 48h: 0.0048±0.00084; PSENEN -/- GG 48 h: 0.0056±0.00094, P=0.0181; PSENEN -/- NT 72 h: 0.0042±0.00056; PSENEN -/- GG 72 h: 0.0051±0.00010; P=0.0238). WT HaCaT cells’ data are not shown since in our experimental conditions, the scratch closed within 24-36 hours. Overall, our results collectively indicate that GG treatment increases cell migration in an in vitro model of HS.
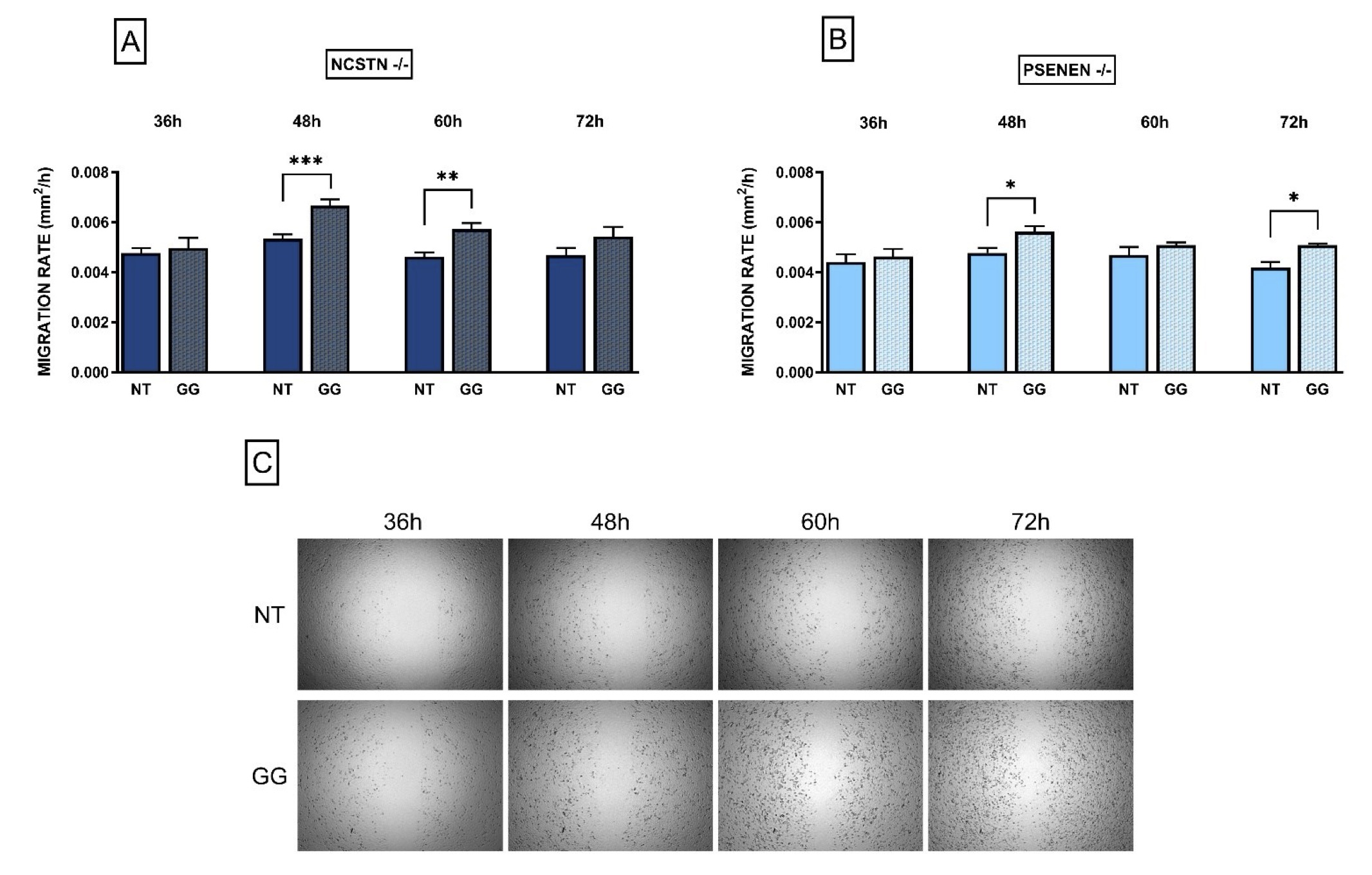
Fig. 3: HaCaT NCSTN -/- and PSENEN -/- migration rate increases after GG treatment. Data are shown as migration rate (mm2/h) with and without GG in both KO cell lines, at 4 different time points (36, 48, 60, 72 hours). (A): Migration rate in NCSTN -/- HaCaT; after 48 and 60 hours from scratch making the difference between treated and non-treated migration rate is statistically significant. P***=0.0002, P**=0.007. (B): Migration rate in PSENEN -/- HaCaT; after 48 and 72 hours from scratch making the difference between treated and non-treated migration rate is statistically significant. P*=0.0181, P*=0.0238. (C): Representative pictures of HaCaT cells during scratch assay at 4 different time points (36, 48, 60, 72 hours); the upper line shows non-treated (NT) cells while the lower line shows cells treated with GG.
GG did not induce an increase in the proliferation rate
Cell proliferation of KO cell lines with and without the GG treatment was monitored in real-time with the xCELLigence Real-Time Cell Analysis (Agilent) system.
First of all, we observed that KO HaCaT cell lines, both PSENEN -/- and NCSTN -/-, showed an increase in the proliferation rate compared to WT HaCaT cells. An example of the variation of the cell index value over time is represented in Fig. 4A.
Following GG treatment, we observed that the general trend for the treated KO cell lines is a decrease in the proliferation rate when compared to non-treated cells. Specifically starting from 48 hours, the difference is statistically significant (Fold change to NT in NCSTN -/- with GG, T48: 0.80±0.056, P=0.0404; T60: 0.66±0.043, P=0.0001; T72: 0.67±0.049, P=0.0001. Fold change to NT in PSENEN -/- with GG, T48: 0.63±0.074, P=0.0032; T60: 0.51±0.067, P=<0.0001; T72: 0.53±0.078, P=<0.0001) (Fig. 4B, C and D).
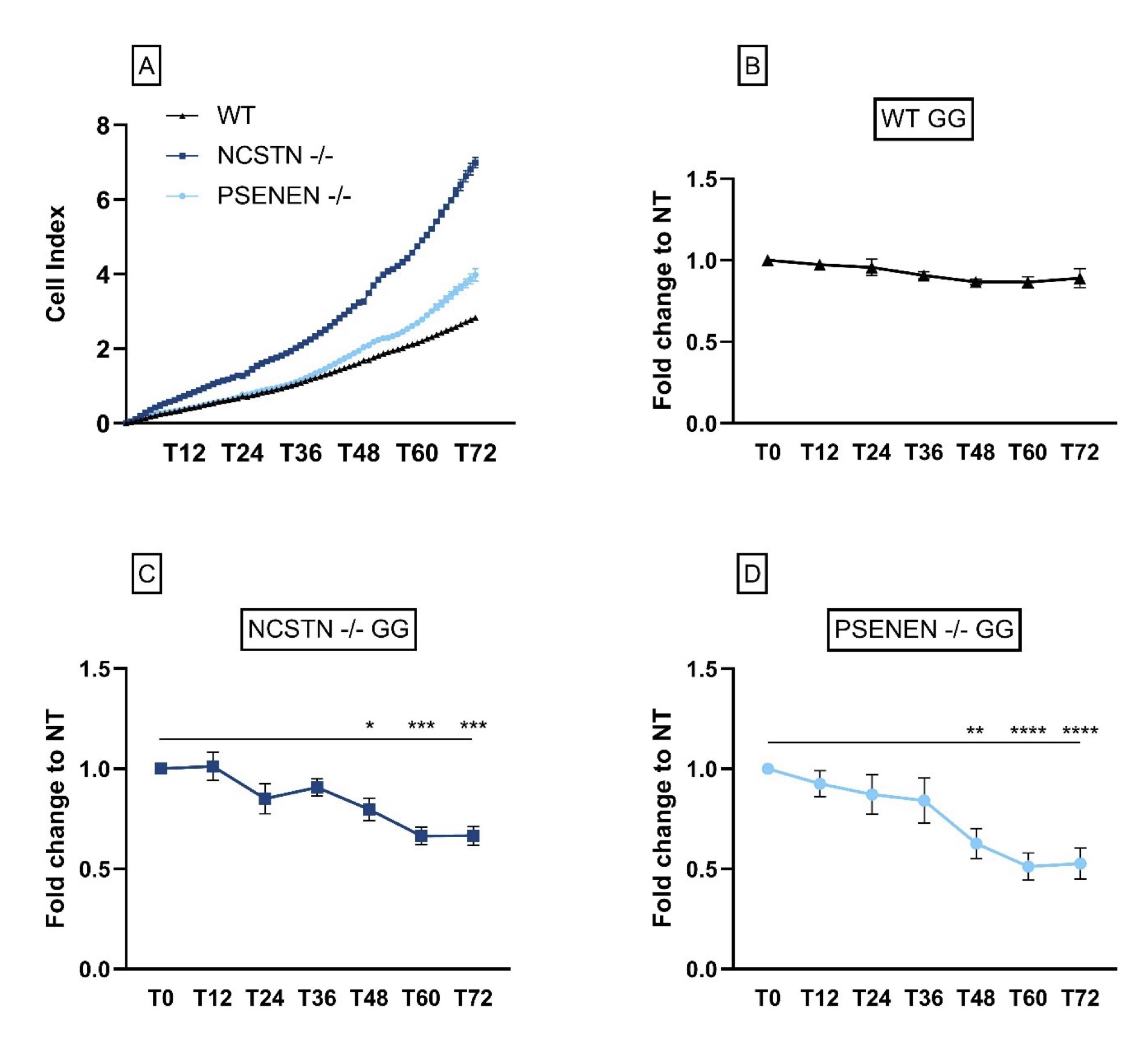
Fig. 4: KO HaCaT cells proliferate at a higher rate when compared to HaCaT WT; GG treatment does not increase proliferation rate. (A): Results from one experiment are shown here as an example; NCSTN -/- (dark blue line), PSENEN -/- (light blue line) and wild-type cells (black line). Data are represented as cell index values of single time points for every cell line. For every cell line, data are shown as fold change of the GG-treated cells relative to the NT at different time points. Fold change of: (B) wild-type HaCaT (C) NCSTN-/- (P*=0.0404, P***=0.0001) and (D) PSENEN-/- (P**=0.0032, P****=<0.0001).
Pathway enrichment analysis in NCSTN -/- cells treated and non-treated with GG
While the findings obtained for NCSTN -/- were straightforward and clear, the ones obtained for WT, as well as PSENEN -/-, were more complex and difficult to interpret clearly. So, we specifically cited only the NCSTN -/- treated and non-treated with GG proteome data. For proteomic analysis, NCSTN -/- HaCaT cells were treated with GG for 24 hours. Comparisons were conducted between GG-treated cells and untreated cells. A total of 5,763 proteins from both samples were quantified using label-free quantification.
The obtained raw abundance data were statistically processed by the DEP R package [20], which included normalization using Variance Stabilizing Transformation, imputation of missing data using random draws from a Gaussian distribution centered around a minimal value (MinProb; q =0.01), and differentially expressed protein analysis using linear models and Empirical Bayes statistics.
Based on the obtained false discovery rate (FDR)-adjusted p-value and fold change, statistically significant changes in protein levels were observed for both conditions. Upregulation and downregulation were based on log2 fold-change >0.58 and log2 fold-change <-0.62, respectively, for genes with FDR adjusted p-value < 0.05. GG increased the expression of 40 proteins and decreased the level of 26 proteins compared to the non-treated cells (Fig. 5, see Supplementary Table 1 for the complete list of all proteins).
Pathway enrichment analysis suggested that these proteins are significantly associated with 22 molecular pathways (entities FDR p-value < 0.05) (Table 1).
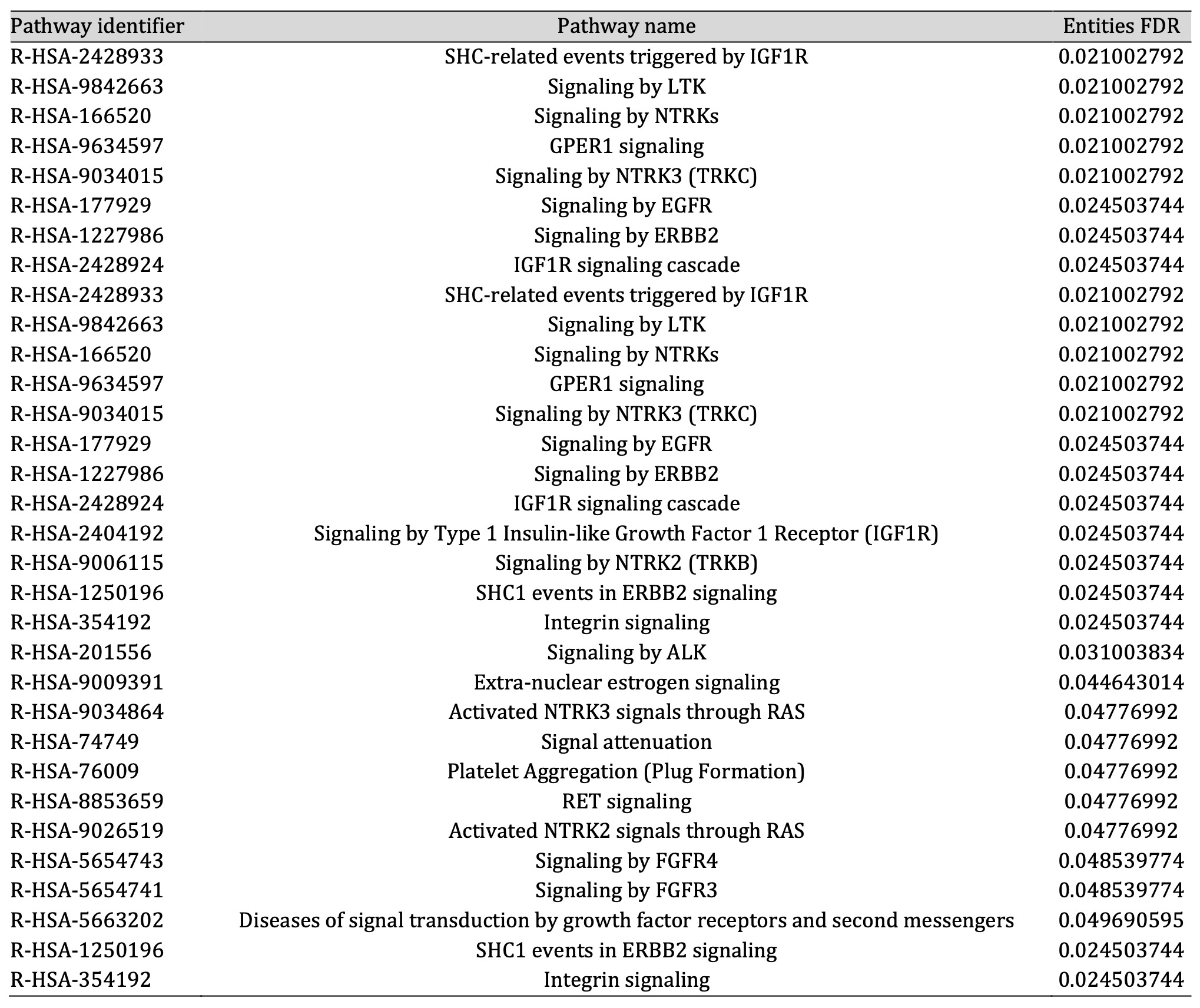
Table 1: All significant pathways obtained by enrichment analysis
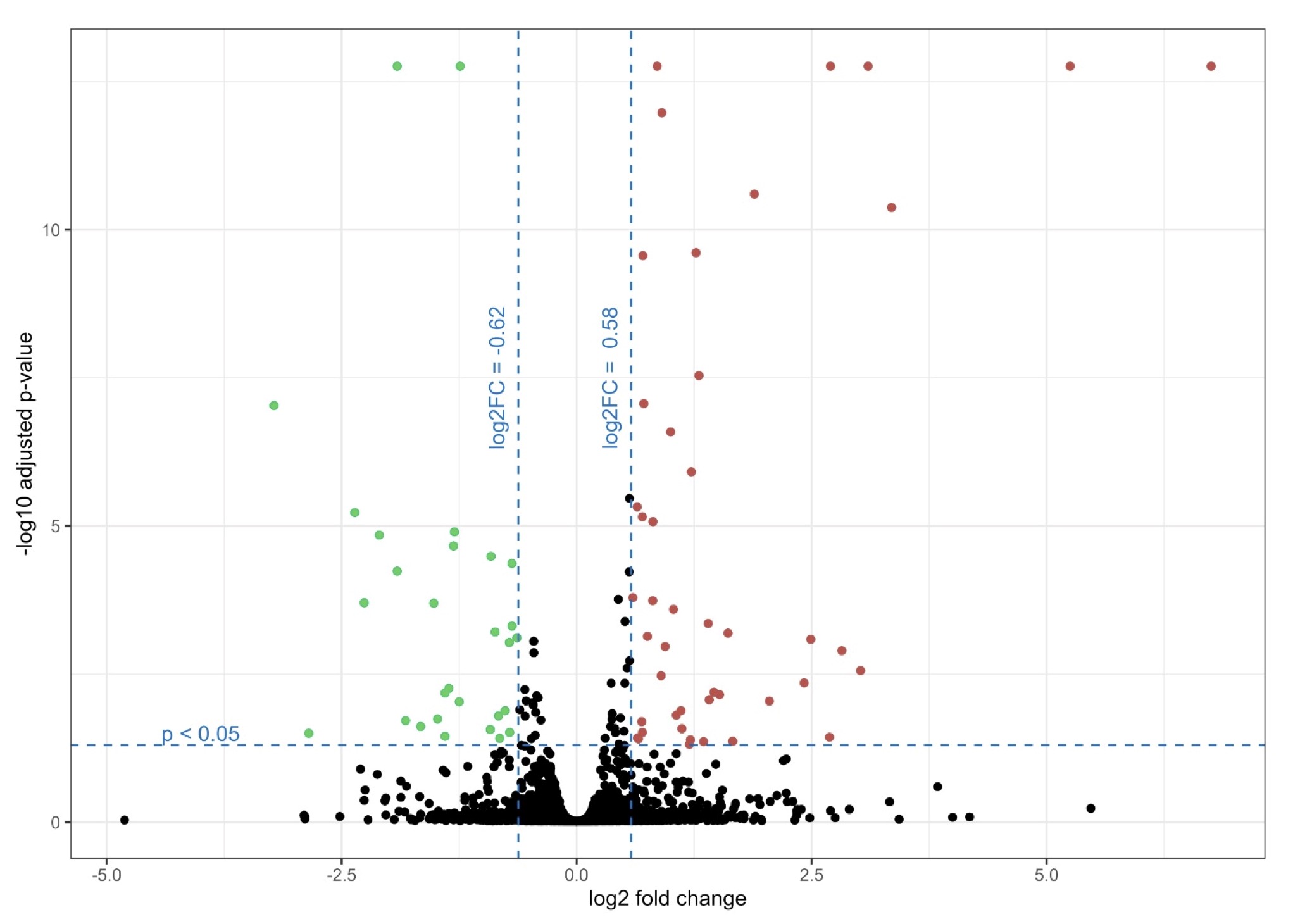
Fig. 5: Volcano plot showing proteomics data of NCSTN -/- HaCaT cells treated with GG compared to NCSTN -/- HaCaT cells without treatment. Volcano plot illustrates the log2 fold change (x axis) and -log10 adjusted p-value (y axis). Green color marks down regulated proteins with log2 fold-change < -0.62 and red color marks upregulated proteins with log2 fold-change > 0.58.
Discussion
Hidradenitis suppurativa (HS) is a chronic, auto-inflammatory and debilitating skin condition of the pilosebaceous unit. Recent findings propose that the pathophysiology of HS appears to be the product of hyperkeratinization, hyperplasia and inflammation caused by genetic predisposition [22, 23]. Despite an abundance of familial cases, NCSTN and PSENEN are the main genes so far known to be majorly mutated in HS; these genes encode for two essential components of the γ-secretase multiprotein complex [24, 25].
All mutations found in both NCSTN and PSENEN genes lead to γ-secretase complex instability and degradation of all its subunits [26, 27]; in fact, NCSTN and PSENEN gene-silenced HaCaT cell model are characterised by an increase in cell proliferation, probably through a modulation of phosphoinositide 3-kinase (PI3K)/AKT pathway and the downregulation of genes involved in the Notch signalling [26, 28].
Considering the pleiotropic role of AQP3-facilitated water and glycerol transport in different keratinocytes’ processes, such as migration and proliferation, and that its dysregulation alters keratinocyte proliferation, skin water loss and decreased skin hydration [4, 5,7], we evaluated the possible pathophysiological involvement of this AQP in in our genetic in vitro HS models, consisting in two lines of immortalised human keratinocyte (HaCaT) knock-out for NCSTN -/- and PSENEN -/-.
We observed a significant decrease of AQP3 gene expression and its protein levels in KO HaCaT cell lines, NCSTN -/- and PSENEN -/- cells, compared to wild-type (WT) cells, thus supporting the hypothesis that low levels of AQP3 could contribute to HS development, consequently suggesting relevance for skin AQP3 as a novel potential actor and therapeutic target in HS.
Previous published data demonstrated that glyceryl glucoside (GG; alpha-D-glucopyranosyl-alpha-(1->2)-glycerol; C9H18O8 ), a glucose derivative, is able to increase AQP3 expression levels [18]. We therefore tested the ability of this promising compound to restore the normal gene expression and protein levels of AQP3 in our in vitro model of HS, confirming that GG is able to significantly increase both mRNA and protein levels in all tested cell lines.
Another crucial aspect to consider is that HS is characterized by hyperplasia of the follicular epithelium. Lin Jin et al. thoroughly observed that HS lesional keratinocytes had enhanced proliferation capacity compared to those from healthy skin [22]. Keratinocyte hyperproliferation in HS has been associated with mutations in γ-secretase genes in familial forms of HS, through an upregulation of phosphorylated Akt (p‐AKT) [29]. These data are in line with our results that show an increase in the proliferation rate of NCSTN -/- and PSENEN -/- compared to WT HaCaT.
Interestingly, GG treatment also induced a significant reduction in hyperproliferation in both NCSTN -/- and PSENEN -/- keratinocytes, contrary to what has been registered in WT HaCaT cells.
According to our proteomic analysis on NCSTN -/- keratinocytes treated and non-treated with GG, being aware of the limitations due to the analyses performed in an in vitro model of the disease, we found two different pathways associated with keratinocyte proliferation. The first is GPER1 (G protein-coupled estrogen receptor 1) signaling (R-HSA-9634597), whose alteration has been detected in skin biopsies of psoriasis lesions when compared to healthy skin and whose dysregulation has been associated with keratinocyte proliferation [30]. The second one is the FGFR4 (Fibroblast Growth Factor Receptor 4) signaling (R-HSA-5654743); interestingly, it has been demonstrated that FGF19 (Fibroblast Growth Factor 19) is able to promote a high proliferation rate in psoriatic keratinocytes through the FGFR4 signaling route [31].
In the context of HS, the processes of wound healing and of resolution of lesions often represent critical aspects for HS patients. Indeed, an increment in keratinocyte migration could be particularly useful to foster these processes [32].
Another key role of AQP3 is to aid epidermal cell migration during the wound healing process. Hara-Chikuma and Verkman observed that keratinocytes isolated from AQP3-deficient mice showed reduced migration compared with cells derived from WT mice. They also confirmed these findings in normal human keratinocytes KO for AQP3 showing decreased glycerol uptake and scratch wound healing [33]. Our data concur with all this previous evidence. In fact, the increase in AQP3 levels, due to GG treatment, leads to an increment in the migration of NCSTN -/- and PSENEN -/- keratinocytes. Noteworthy, in NCSTN -/- keratinocytes, treated and non-treated with GG, the dysregulation of the integrin signaling pathway (R-HSA-354192) was statistically significant. Integrins are transmembrane receptors that upon ligand binding activate several signal transduction pathways. Specifically, in the epidermis they are mainly involved in extracellular matrix binding, therefore in migration and wound healing events [34].
Lastly, it is worth focusing the attention on other two pathways found to be significantly unbalanced: EGFR (Epidermal growth factor receptor) (R-HSA-177929) and ERBB2 (Erb-B2 Receptor Tyrosine Kinase 2) signaling (R-HSA-1227986). EGFR is widely known to impact several keratinocytes biological processes including proliferation, adhesion and migration [35]. Likewise ERBB2, also known as Human Epidermal Growth Factor Receptor 2 (HER2), is a member of the EGF family of receptors and seems to be able to regulate the process of proliferation and migration [36].
Altogether, considering the fact that these pathways were altered in GG-treated NCSTN -/- HaCaT cells when compared to NCSTN -/- non-treated cells, and that their involvement in multiple keratinocyte processes is fundamental for skin homeostasis, these data strongly corroborate the importance of the GG treatment in HaCat cells in which NCSTN gene has been knocked out.
Hara-Chikuma et al., in their study conducted in AQP3-deficient mice, proposed a mechanism for AQP3-dependent wound healing involving AQP3-facilitated water and glycerol transport. In detail they hypothesised that the glycerol transport by AQP3 is important for the proliferation of epidermal cells, while water transport is important for epidermal cell migration. They also observed that a supplementation of Glycerol corrected the impairment in wound repair and cell proliferation in AQP3-deficient mice [33]. In this case, the correction obtained after Glycerol treatment is not correlated with the alteration of AQP3 expression, as it can be observed in our case.
It must also be considered that AQP3 is also peroxiporin capable of facilitating the transport of H2O2 across the membrane. In recent years there has been convincing evidence indicating the relevance of AQP3-mediated H2O2 import in cell migration [37, 38].
Kyoko Nakahigashi et al., found an upregulation of AQP3 in atopic dermatitis mouse models that is involved in keratinocyte proliferation and epidermal hyperplasia. In this case they also suggest that AQP3 inhibition may be beneficial for the treatment of atopic dermatitis [8].
In our case, we found a decrease of the levels of AQP3 gene and protein in our genetic HS in vitro model. The subsequent increment of AQP3 levels, induced by GG treatment, caused an increase in cell migration and also a decrease in the hyperproliferation rate. The increase in cell migration is in line with literature data, partly discussed previously, that show a close correlation between keratinocyte migration and AQP3 level. On the contrary, the different action found in the increase of AQP3 level in the hyperproliferation rate, requires further studies.
Conclusion
Our findings reveal that Glyceryl Glucoside (GG) can significantly increase both mRNA and protein levels of Aquaporin-3 (AQP3) in all tested cell lines. However, it is important to note that these results were obtained from an in vitro model, which only partially mimics the complex etiology and pathogenesis of Hidradenitis Suppurativa (HS). We acknowledge the limitations of our in vitro model, as it may not fully capture the intricate interactions and environment present in vivo . To validate our hypothesis regarding the involvement of AQP3 in Hidradenitis Suppurativa and the therapeutic potential of GG, it is essential to conduct in vivo studies. Such studies would provide a more comprehensive understanding of how AQP3 dysregulation contributes to HS and confirm whether GG can effectively restore AQP3 levels and alleviate the disease in a living organism.
To our knowledge, this is the first report demonstrating a dysregulation of AQP3 in an in vitro model that mimics genetic-driven HS. Based on our findings, changes in the expression levels of AQP3 may play a significant role in the pathogenesis of Hidradenitis Suppurativa, particularly in cases driven by genetic factors. This assertion is supported by experiments conducted on NCSTN and PSENEN knockout (KO) HaCat cell lines, which illustrate how genetic modifications can influence AQP3 levels and potentially contribute to the development of HS. Targeting this protein could therefore help improve altered processes of keratinocytes that underlie this complex disease, especially those related to migration and proliferation.
In conclusion, our research indicates that AQP3 may be a promising new target for treating Hidradenitis Suppurativa. This discovery paves the way for further research into its potential therapeutic applications. Additionally, given GG’s capability to enhance wound healing in HS, it could serve as a beneficial adjunctive therapy for patients with this condition.
Acknowledgements
Glycoin® natural (GG) reagent was donated by Bitop. This work was supported by a Starting Grant (SG-2019- 12369421) founded by the Italian Ministry of Health.
Author Contributions
CVD and PMT conceived and planned the experiments; CN-M and MB provided the KO cell line; CVD, RG and PMT performed all experiments; CVD, EMN, ES and BM performed proteomics analysis; RM perform bioinformatic analysis; RG, DM, MB, GC, contributed to the interpretation of the results; CVD, EMN, SC, PMT wrote the original draft manuscript; CM, AVM, SC reviewed the manuscript; all authors contributed to the article and approved the submitted version.
Funding Sources
This work was supported by the Italian Ministry of Health, through the contribution given to the Institute for Maternal and Child Health IRCCS Burlo Garofolo, Trieste – Italy (RC16/18).
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors have no conflicts of interest to declare.
References
| 1 | Pimpão C, Wragg D, Da Silva IV, Casini A, Soveral G. Aquaglyceroporin Modulators as Emergent Pharmacological Molecules for Human Diseases. Front Mol Biosci 2022;9:845237.
https://doi.org/10.3389/fmolb.2022.845237 |
| 2 | Tamma G, Valenti G, Grossini E, Donnini S, Marino A, Marinelli RA, et al. Aquaporin Membrane Channels in Oxidative Stress, Cell Signaling, and Aging: Recent Advances and Research Trends. Oxid Med Cell Longev 2018;2018:1-14.
https://doi.org/10.1155/2018/1501847 |
| 3 | Sonntag Y, Gena P, Maggio A, Singh T, Artner I, Oklinski MK, et al. Identification and characterization of potent and selective aquaporin-3 and aquaporin-7 inhibitors. J Biol Chem 2019;294:7377-7387.
https://doi.org/10.1074/jbc.RA118.006083 |
| 4 | Bollag WB, Aitkens L, White J, Hyndman KA. Aquaporin-3 in the epidermis: more than skin deep. Am J Physiol Cell Physiol 2020;318:1144-1153.
https://doi.org/10.1152/ajpcell.00075.2020 |
| 5 | Hara-Chikuma M, Verkman AS. Roles of Aquaporin-3 in the Epidermis. J Invest Dermatol 2008;128:2145-2151.
https://doi.org/10.1038/jid.2008.70 |
| 6 | Tricarico PM, Mentino D, De Marco A, Del Vecchio C, Garra S, Cazzato G, et al. Aquaporins Are One of the Critical Factors in the Disruption of the Skin Barrier in Inflammatory Skin Diseases. Int J Mol Sci 2022;23:4020.
https://doi.org/10.3390/ijms23074020 |
| 7 | Lee Y, Je Y-J, Lee S-S, Li ZJ, Choi D-K, Kwon Y-B, et al. Changes in Transepidermal Water Loss and Skin Hydration according to Expression of Aquaporin-3 in Psoriasis. Ann Dermatol 2012;24:168-174.
https://doi.org/10.5021/ad.2012.24.2.168 |
| 8 | Nakahigashi K, Kabashima K, Ikoma A, Verkman AS, Miyachi Y, Hara-Chikuma M. Upregulation of Aquaporin-3 Is Involved in Keratinocyte Proliferation and Epidermal Hyperplasia. J Invest Dermatol 2011;131:865-873.
https://doi.org/10.1038/jid.2010.395 |
| 9 | Chen M, Peng Q, Tan Z, Xu S, Wang Y, Wu A, et al. Targeting Aquaporin-3 Attenuates Skin Inflammation in Rosacea. Int J Biol Sci 2023;19:5160-5173.
https://doi.org/10.7150/ijbs.86207 |
| 10 | Von Der Werth J, Williams H. The natural history of hidradenitis suppurativa. J Eur Acad Dermatol Venereol 2000;14:389-392.
https://doi.org/10.1046/j.1468-3083.2000.00087.x |
| 11 | De Vita V, McGonagle D. Hidradenitis suppurativa as an autoinflammatory keratinization disease. J Allergy Clin Immunol 2018;141:1953.
https://doi.org/10.1016/j.jaci.2018.01.010 |
| 12 | Jemec GBE. Hidradenitis Suppurativa. N Engl J Med 2012;366:158-164.
https://doi.org/10.1056/NEJMcp1014163 |
| 13 | Sabat R, Jemec GBE, Matusiak Ł, Kimball AB, Prens E, Wolk K. Hidradenitis suppurativa. Nat Rev Dis Primer 2020;6:18.
https://doi.org/10.1038/s41572-020-0149-1 |
| 14 | Wang B, Yang W, Wen W, Sun J, Su B, Liu B, et al. γ-Secretase Gene Mutations in Familial Acne Inversa. Science 2010;330:1065-1065.
https://doi.org/10.1126/science.1196284 |
| 15 | Napolitano M, Megna M, Timoshchuk E, Patruno C, Balato N, Fabbrocini G, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115.
https://doi.org/10.2147/CCID.S111019 |
| 16 | Prens LM, Huizinga J, Janse IC, Horváth B. Surgical outcomes and the impact of major surgery on quality of life, activity impairment and sexual health in hidradenitis suppurativa patients: a prospective single centre study. J Eur Acad Dermatol Venereol 2019;33:1941-1946.
https://doi.org/10.1111/jdv.15706 |
| 17 | Anduquia-Garay F, Rodríguez-Gutiérrez MM, Poveda-Castillo IT, Valdes-Moreno PL, Agudelo-Rios DA, Benavides-Moreno JS, et al. Hidradenitis suppurativa: Basic considerations for its approach: A narrative review. Ann Med Surg 2021;68. DOI: 10.1016/j.amsu.2021.102679
https://doi.org/10.1016/j.amsu.2021.102679 |
| 18 | Schrader A, Siefken W, Kueper T, Breitenbach U, Gatermann C, Sperling G, et al. Effects of Glyceryl Glucoside on AQP3 Expression, Barrier Function and Hydration of Human Skin. Skin Pharmacol Physiol 2012;25:192-199.
https://doi.org/10.1159/000338190 |
| 19 | Tricarico PM, Zupin L, Ottaviani G, Pacor S, Jean-Louis F, Boniotto M, et al. Photobiomodulation therapy promotes in vitro wound healing in nicastrin KO HaCaT cells. J Biophotonics 2018;11:e201800174.
https://doi.org/10.1002/jbio.201800174 |
| 20 | Zhang X, Smits AH, Van Tilburg GB, Ovaa H, Huber W, Vermeulen M. Proteome-wide identification of ubiquitin interactions using UbIA-MS. Nat Protoc 2018;13:530-550.
https://doi.org/10.1038/nprot.2017.147 |
| 21 | Yu G, He Q-Y. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol Biosyst 2016;12:477-479.
https://doi.org/10.1039/C5MB00663E |
| 22 | Jin L, Kashyap MP, Chen Y, Khan J, Guo Y, Chen JQ, et al. Mechanism underlying follicular hyperproliferation and oncogenesis in hidradenitis suppurativa. iScience 2023;26:106896.
https://doi.org/10.1016/j.isci.2023.106896 |
| 23 | Jones D, Banerjee A, Berger PZ, Gross A, McNish S, Amdur R, et al. Inherent differences in keratinocyte function in hidradenitis suppurativa: Evidence for the role of IL-22 in disease pathogenesis. Immunol Invest 2018;47:57-70.
https://doi.org/10.1080/08820139.2017.1377227 |
| 24 | Moltrasio C, Tricarico PM, Romagnuolo M, Marzano AV, Crovella S. Hidradenitis Suppurativa: A Perspective on Genetic Factors Involved in the Disease. Biomedicines 2022;10:2039.
https://doi.org/10.3390/biomedicines10082039 |
| 25 | Pace NP, Mintoff D, Borg I. The Genomic Architecture of Hidradenitis Suppurativa-A Systematic Review. Front Genet 2022;13:861241.
https://doi.org/10.3389/fgene.2022.861241 |
| 26 | Xiao X, He Y, Li C, Zhang X, Xu H, Wang B. Nicastrin mutations in familial acne inversa impact keratinocyte proliferation and differentiation through the Notch and phosphoinositide 3‐kinase/AKT signalling pathways. Br J Dermatol 2016;174:522-532.
https://doi.org/10.1111/bjd.14223 |
| 27 | Garcovich S, Tricarico PM, Nait‐Meddour C, Giovanardi G, Peris K, Crovella S, et al. Novel nicastrin mutation in hidradenitis suppurativa-Dowling-Degos disease clinical phenotype: more than just clinical overlap? Br J Dermatol 2020;183:758-759.
https://doi.org/10.1111/bjd.19121 |
| 28 | Li W, Zhang Y, Jia W, He Y, Xu H, Lin L, et al. Effect of PSENEN gene silencing on the proliferation of and γ-secretase expression in HaCaT cells. Chin J Dermatol 2021;318-324.
|
| 29 | He Y, Li C, Xu H, Duan Z, Liu Y, Zeng R, et al. AKT‐dependent hyperproliferation of keratinocytes in familial hidradenitis suppurativa with a NCSTN mutation: a potential role of defective miR‐100‐5p. Br J Dermatol 2020;182:500-502.
https://doi.org/10.1111/bjd.18460 |
| 30 | Pérez-Escudero N, Cabas I, Cantón-Sandoval J, Carrión MB-, García-Moreno D, Mulero V, et al. The estrogen receptor GPER1 attenuates skin inflammation via inhibition of keratinocyte hyperproliferation. Dev Comp Immunol 2023;148:104992.
https://doi.org/10.1016/j.dci.2023.104992 |
| 31 | Yu X, Yan N, Li Z, Hua Y, Chen W. FGF19 sustains the high proliferative ability of keratinocytes in psoriasis through the regulation of Wnt/ GSK ‐3β/β‐catenin signalling via FGFR 4. Clin Exp Pharmacol Physiol 2019;46:761-769.
https://doi.org/10.1111/1440-1681.13103 |
| 32 | Mayur O, Vedak P. Wound care in hidradenitis suppurativa. Dermatol Rev 2022;3:150-153.
https://doi.org/10.1002/der2.127 |
| 33 | Hara-Chikuma M, Verkman AS. Aquaporin-3 facilitates epidermal cell migration and proliferation during wound healing. J Mol Med 2008;86:221-231.
https://doi.org/10.1007/s00109-007-0272-4 |
| 34 | Longmate WM, DiPersio CM. Integrin Regulation of Epidermal Functions in Wounds. Adv Wound Care 2014;3:229-246.
https://doi.org/10.1089/wound.2013.0516 |
| 35 | Tran QT, Kennedy LH, Leon Carrion S, Bodreddigari S, Goodwin SB, Sutter CH, et al. EGFR regulation of epidermal barrier function. Physiol Genomics 2012;44:455-469.
https://doi.org/10.1152/physiolgenomics.00176.2011 |
| 36 | Dahlhoff M, Zouboulis CC, Schneider MR. Expression of dermcidin in sebocytes supports a role for sebum in the constitutive innate defense of human skin. J Dermatol Sci 2016;81:124-126.
https://doi.org/10.1016/j.jdermsci.2015.11.013 |
| 37 | Miller EW, Dickinson BC, Chang CJ. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc Natl Acad Sci 2010;107:15681-15686.
https://doi.org/10.1073/pnas.1005776107 |
| 38 | Hara-Chikuma M, Chikuma S, Sugiyama Y, Kabashima K, Verkman AS, Inoue S, et al. Chemokine-dependent T cell migration requires aquaporin-3-mediated hydrogen peroxide uptake. J Exp Med 2012;209:1743-1752.
https://doi.org/10.1084/jem.20112398 |