Nitric Oxide-Dependent Regulation of Oxygen-Related Processes in a Rat Model of Lead Neurotoxicity: Influence of the Hypoxia Resistance Factor
bNicolaus Copernicus University in Toruń, Collegium Medicum in Bydgoszcz, Department of Medical Biology and Biochemistry, Department of Ecology and Environmental Protection, M. Skłodowska-Curie St. 9, 85-094 Bydgoszcz, Poland,
cUniversity of Zielona Góra, Faculty of Biological Sciences, Institute of Biological Sciences, Department of Biotechnology, Prof. Z. Szafran St. 1, 65-516 Zielona Góra, Poland,
dDepartment of Ecology, Geography and Nature Management, T.H. Shevchenko National University “Chernihiv Colehium”, Hetmana Polubotka St. 53, 14013 Chernihiv, Ukraine
Keywords
Abstract
Background/Aims:
Lead exposure is known to induce oxidative stress and neurotoxicity. Nitric oxide (NO) plays an important role in modulating oxidative stress, with L-arginine as a precursor of NO and Nω-nitro-L-arginine (L-NNA) as an inhibitor of NO synthase, an enzyme that catalyses the production of nitric oxide (NO) from L-arginine.Methods:
This study investigated the differential effects of L-arginine and L-NNA on markers of oxidative stress and biochemical changes in brain tissue from rats with different levels of resistance to hypoxia exposed to lead nitrate. Rats with either low or high resistance to hypoxia were exposed to lead nitrate (oral 3.6 mg lead nitrate/kg b.w. per day for 30 days) and treated with L-arginine (600 mg/kg b.w., i.p., 30 min before and after exposure to lead nitrate) or L-NNA (35 mg/kg b.w., i.p., 30 min before and after exposure to lead nitrate). Brain tissue samples were analysed for lipid peroxidation, oxidative modification of proteins, and activity of antioxidant enzymes, including superoxide dismutase, catalase, glutathione reductase, and peroxidase, and total antioxidant status (TAS). We also examined the biomarkers of biochemical pathways involving the activity of alanine and aspartate aminotransferases, succinate dehydrogenase (SDH), and α-ketoglutarate dehydrogenase (KGDH). In addition, the trend observed was supported by assessments of the acetylcholine levels and acetylcholinesterase activity (ACh-AChE system) in brain tissue.Results:
In rats with low resistance to hypoxia, the L-arginine treatment significantly reduced lipid peroxidation and oxidative protein modification but increased antioxidant enzyme activity, suggesting a protective effect against lead-induced oxidative stress. Conversely, in rats with high resistance to hypoxia, L-NNA had a protective effect, reducing lead-induced oxidative damage and decreasing lipid peroxidation, whereas L-arginine exacerbated oxidative stress and impaired antioxidant defences. These findings were supported by corresponding changes in the acetylcholine-acetylcholinesterase system, reflecting the observed patterns of lead-induced oxidative stress and neurotoxicity. The study shows that L-arginine exerts a protective effect by reducing lead-induced oxidative damage via an improvement in TAS. Our study shows that lead nitrate exposure significantly increases ala-nine and aspartate aminotransferase activity in brain tissue, with L-arginine exacerbating and L-NNA reversing this effect. The lead nitrate exposure also affected the activities of SDH and KGDH, which are important for cellular energy production and hypoxia resistance, with L-arginine altering SDH activity depending on the level of resistance and L-NNA enhancing both SDH and KGDH activities. These trends were further validated by alterations in the ACh-AChE system, highlighting the differential role of NO-dependent mechanisms in modulating lead-induced neurotoxicity based on hypoxia resistance.Conclusion:
These findings suggest potential targeted therapeutic strategies based on the oxidative stress profile and highlight the potential of nitric oxide system modulators in counteracting lead-induced biochemical alterations and the dynamics of the ACh-AChE system depending on the individual physiological reactivity of organisms.Introduction
Neurotoxicity refers to the neurophysiological changes caused by exposure to toxic agents, including heavy metals, pharmaceuticals, organophosphates, and bacterial and animal neurotoxins, which can lead to neurocognitive symptoms and psychiatric disorders [1, 2]. Among these, lead (Pb) exposure is particularly prevalent and has a significant impact on neuropsychological and functional health due to its effects on cognitive and psychological functions, including intelligence, memory, executive function, attention, processing speed, language, visuospatial skills, motor skills, and mood [3, 4, 5]. Lead exposure is associated with a range of adverse effects, including cognitive impairment, developmental delay in children, and neurodegenerative diseases in adults. These health concerns highlight the continued need for research and intervention strategies to address lead contamination and mitigate its long-term effects [6].
Studies highlight that lead exposure poses serious health risks, including cognitive impairment and severe neurophysiological changes, especially in vulnerable populations, such as children and those with prolonged exposure [7, 8]. Several comprehensive studies have highlighted the critical impact of Pb on neurological health and the importance of addressing its toxic effects through continued research and public health efforts [4, 9, 10]. Lead is a widespread and highly potent neurotoxicant that affects a range of neurophysiological and behavioural functions, with the primary effect exerted on the central nervous system (CNS), as shown in Fig. 1. This provides the basis for understanding the broader effects of Pb exposure on brain health and general neurofunction [11]. In adults, lead poisoning can lead to encephalopathy, characterised by such symptoms as insomnia, poor attention span, vomiting, convulsions and coma, which may indicate severe neurotoxic effects with potentially life-threatening consequences [12].
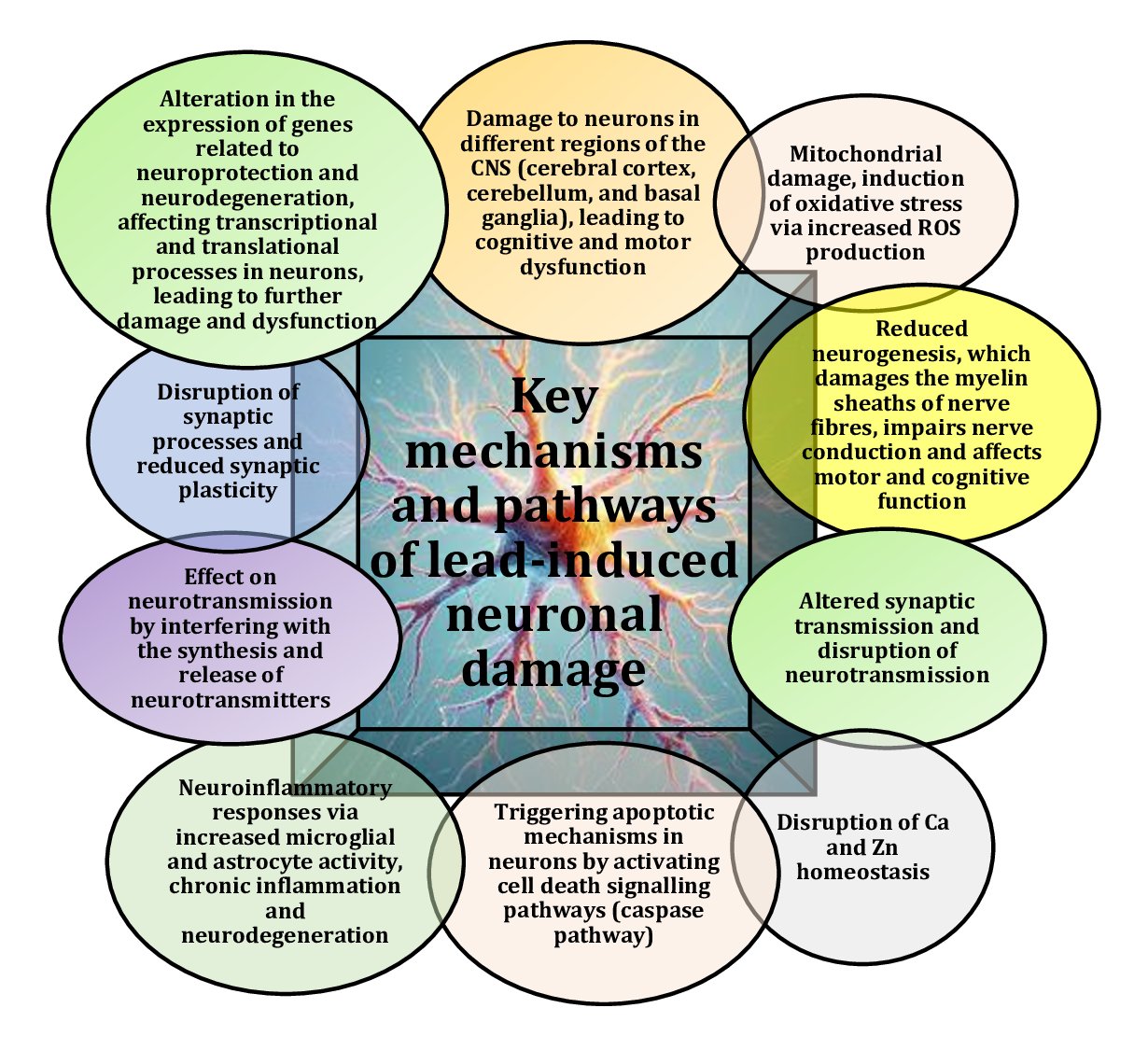
Fig. 1: Key mechanisms and pathways of lead-induced neuronal damage [1-12].
Pb affects the nitric oxide (NO) system in brain tissue primarily by inhibiting nitric oxide synthase (NOS), resulting in reduced NO levels [12]. Pb can directly inhibit the activity of NOS, an enzyme critical for NO production, thereby disrupting normal neurotransmission and regulation of cerebral blood flow [13]. A reduction in the expression of neuronal nitric oxide synthase (nNOS), endothelial nitric oxide synthase (eNOS), and inducible nitric oxide synthase (iNOS) was observed, suggesting a broad effect of Pb on nitric oxide signalling, with impairment affecting various physiological processes regulated by these isoforms, including neurotransmission, vascular function, and immune responses [14, 15]. In addition, lead reduces the bioavailability of NO by increasing the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS), which can react with NO to form toxic products, such as peroxynitrite. These reactions reduce the effective concentration of NO in brain tissue [16]. The interactions between the NO system and the effects of lead are shown in Fig. 2.
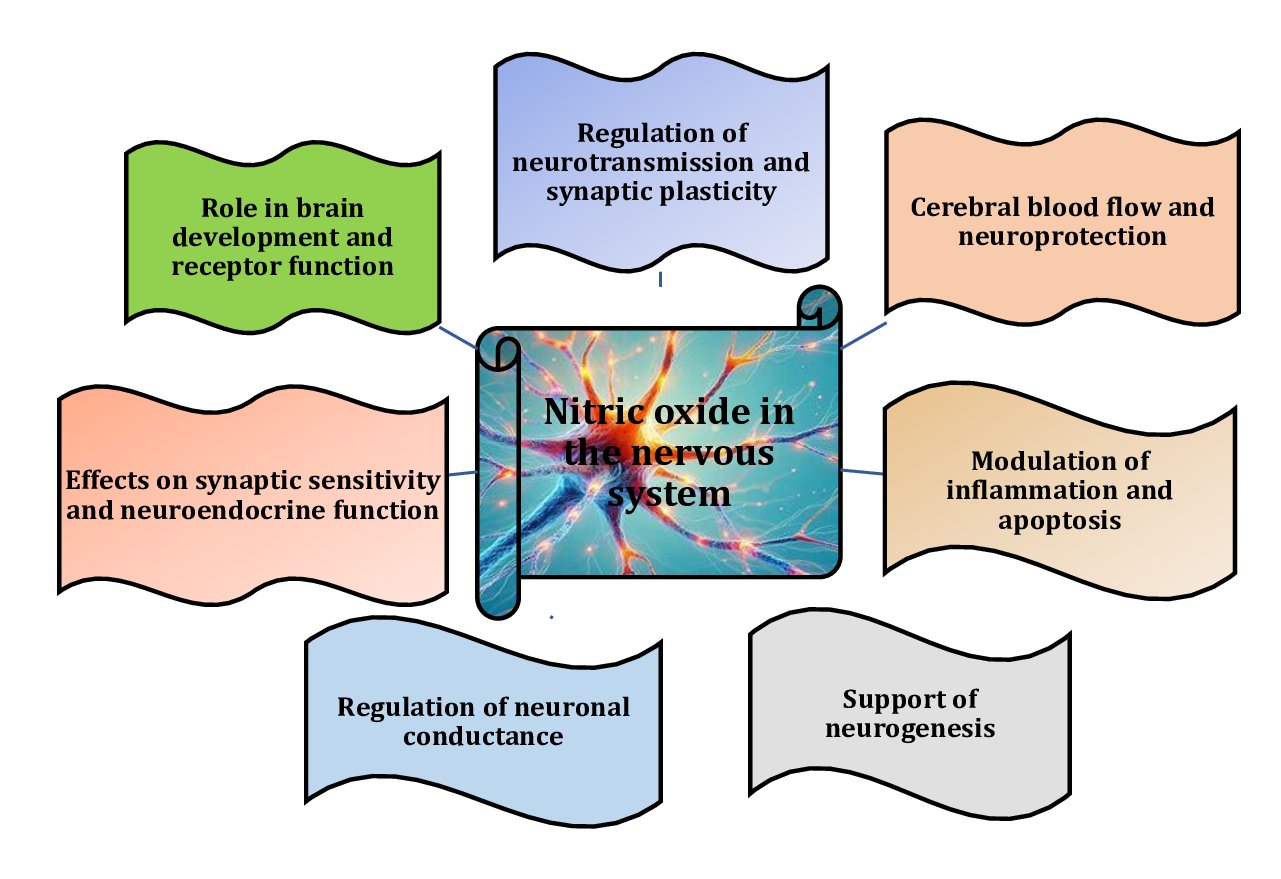
Fig. 2: Role and importance of nitric oxide in the nervous system [12-16].
Chetty et al. [15] demonstrated the effects of environmental Pb exposure on the developing nervous system, in particular its role in causing cognitive deficits in young organisms, focusing on the activity of neuronal nitric oxide synthase (nNOS) in the cerebellum and hippocampus of developing rat brains. A study conducted by Reddy et al. [7] also confirmed that exposure to low levels of Pb during early development is associated with behavioural abnormalities and cognitive deficits in children. They found that Pb exposure led to a reduction in both the activity and protein levels of nNOS in these regions, and this reduction is significant because nitric oxide, a key biological messenger, regulates important neurophysiological processes, including long-term potentiation, which is essential for learning and memory. The reduced nNOS activity and the subsequent decrease in NO production may contribute to cognitive deficits associated with Pb exposure, highlighting the adverse effects of disruptions in NO signalling on brain development and cognitive function. In children, Pb-induced encephalopathy manifests as mental dullness, vomiting, irritability, and anorexia, and long-term exposure results in reduced cognitive function and mental deficits, illustrating the severe effects of Pb on the developing brain [17]. Pb exposure has been shown to have significant effects on cognitive function, particularly through its effects on oxidative stress, the blood-brain barrier, Ca²⁺-dependent processes, and nitric oxide production, and these mechanisms collectively contribute to Pb-induced neurotoxicity [18, 19].
Oxidative modifications of proteins are of key importance in the context of lead-induced neurotoxicity due to their significant impact on neuronal health [19]. These modifications can lead to increased protein aggregation, causing proteins to denature and form toxic aggregates that disrupt neuronal function and contribute to neurodegeneration [20-22]. Lead is known to disrupt calcium (Ca²⁺)-dependent processes by interfering with the function of calcium channels, thereby affecting Ca²⁺ homeostasis within cells [23]. As NO acts as a regulator of Ca²⁺-dependent processes, changes in Ca²⁺ levels may further affect NO function [24]. Lead can also activate inflammatory pathways, leading to increased levels of pro-inflammatory cytokines, which can affect NOS expression and NO production both directly and indirectly through oxidative stress [25]. In addition, oxidative stress can alter cellular signalling pathways, including those involved in neuroplasticity, leading to impairments in cognitive and motor function [26]. Understanding these processes is essential for developing strategies to mitigate the neurotoxic effects of lead and other stressors on neuronal cells [19]. The oxidative modification of proteins plays a critical role in lead-induced neurotoxicity, as this process profoundly affects neuronal protein structure and function, leading to significant neuronal damage through such mechanisms as increased protein aggregation, loss of protein function, and activation of apoptotic pathways [22]. Understanding these effects is essential to elucidate the broader implications of oxidative stress in neurotoxic conditions and to develop potential therapeutic strategies [16].
Acetylcholine (ACh) plays a critical role in the regulation of cerebral blood flow by activating M5 muscarinic receptors on endothelial cells, triggering calcium signalling that leads to the production of NO, a potent vasodilator [27, 28]. This process ensures adequate blood flow to the brain, particularly the cortex, supporting neuronal activity and energy metabolism. The acetylcholine-NO pathway is particularly important in lead poisoning, where both endothelial function and neurotransmitter signalling are disrupted [29]. Lead exposure can impair NO production, reducing cerebral blood flow and contributing to neurological deficits such as cognitive impairment [30]. Understanding this pathway may provide therapeutic insights to mitigate the vascular and neurotoxic effects of lead poisoning by improving endothelial function and NO production [31]. Research suggests that ACh plays a critical role in the regulation of cerebral blood flow through an NO-dependent mechanism [32]. Synaptic release of acetylcholine from neurons activates muscarinic M5 receptors (M5-mAchRs) on brain microvascular endothelial cells [28]. These receptors trigger the Ca2+ signalling pathway, which is essential for the synthesis and release of NO. NO is a potent gasotransmitter that causes vasodilation, increasing blood flow to specific regions of the brain. This process is particularly important for the cerebral cortex, where adequate blood flow is required to maintain neuronal activity and energy metabolism [13, 33]. Several studies have examined different aspects of the neurotransmitter systems affected by Pb exposure. For example, some studies have found significant decreases in synaptosomal acetylcholinesterase (AChE) and mitochondrial monoamine oxidase (MAO) activities as well as increased levels of ACh, dopamine, and epinephrine [34]. These results differ from those found in other studies, which may be due to differences in the experimental design, including the timing and duration of exposure [35, 36].
Through key intermediates, such as glutamate, α-ketoglutarate, succinate, and oxaloacetate, which integrate amino acid metabolism with the citric acid cycle (Krebs cycle), pathways involving alanine and aspartate aminotransferases, succinate dehydrogenase, and α-ketoglutarate dehydrogenase are interconnected [37, 38]. Glutamate produced by transaminases is a precursor of α-ketoglutarate, a critical intermediate in the Krebs cycle. α-Ketoglutarate is then converted by α-Ketoglutarate dehydrogenase to succinyl-CoA, which generates NADH for ATP production. Succinate, formed from α-ketoglutarate, is used by succinate dehydrogenase, linking the Krebs cycle to the electron transport chain [39]. Oxaloacetate, produced by aspartate aminotransferase, helps to restart the Krebs cycle by condensing with acetyl-CoA, ensuring continuous energy production essential for brain function. The links between Krebs cycle function and the nitric oxide system under different functional loads, depending on individual hypoxia tolerance, have been discussed in several studies [40, 41]. These studies investigate how these pathways interact to maintain cellular energy balance and support physiological adaptation in response to hypoxic conditions. The research highlights the importance of individual resistance to hypoxia in modulating these metabolic processes, thereby influencing overall organismal tolerance to hypoxia and stress [42, 43].
Research [40, 41] highlights the importance of understanding the impact of L-arginine, which stimulates nitric oxide biosynthesis, on stress conditions. By exploring the mechanisms by which L-arginine affects NO production, these studies shed light on its role in various physiological functions and the effect of individual differences in physiological reactivity on organism responses to stress and pathological processes [42]. This understanding is critical for the development of more effective and personalised therapeutic and preventive strategies [42]. Studies of individual susceptibility to lead toxicity, particularly in relation to the role of nitric oxide signalling, are a critical area of research that has not been extensively explored in the existing literature [44]. Nitric oxide plays an important role in several physiological processes, including neuronal signalling and immune responses [45]. Variation in individual responses to lead exposure, potentially influenced by differences in NO metabolism and signalling, may have important implications for understanding the differential effects of lead toxicity in populations. Research in this area can provide new insights into the contribution of genetic and biochemical factors to susceptibility and resistance to lead-induced damage.
This study was a continuation of our previous research [41] in which we investigated the effects of the nitric oxide system on lead nitrate exposure using liver tissue as the primary model. Although our previous work provided valuable insights into these interactions, it did not fully address all relevant aspects of this phenomenon. In particular, we did not explore the unique physiological characteristics of nervous tissue, which differ significantly from those of liver tissue. Given these differences, it was essential to investigate the effect of modulation of the nitric oxide system via the nitric oxide precursor amino acid L-arginine or the nitric oxide synthase inhibitor Nω-nitro-L-arginine (L-NNA) administered in vivo on brain tissue in the context of lead poisoning. In addition, this study aimed to investigate the individual physiological characteristics of nervous tissue during oxidative stress damage, in particular its basic physiological response characteristics, which we correlated with resistance to hypoxia. By focusing on these aspects, we sought to gain a more comprehensive understanding of the effects of lead exposure on different tissue types and their associated individual responses to hypoxic conditions. Investigating the role of nitric oxide in modulating individual responses to lead exposure is particularly important because it may help identify biomarkers or therapeutic targets for personalised therapeutic management. By understanding the influence of NO-related pathways on lead toxicity, researchers may be able to develop more targeted and effective interventions. This knowledge could lead to improved prevention and treatment strategies tailored to the specific susceptibilities of individuals based on their nitric oxide profiles. Our study highlights the effects of orally administered lead, and the inclusion of L-arginine in the diet is an important consideration for individuals at risk of lead exposure. By investigating the impact of L-arginine on lead-induced toxicity, we can better understand its potential role in mitigating oxidative damage and enhancing overall cellular defence mechanisms. This knowledge could inform dietary recommendations and therapeutic approaches aimed at minimising the adverse effects of lead, particularly in populations at significant risk of exposure.
The aim of this study was to assess the effects of oral lead nitrate exposure on various aspects of oxidative and energetic processes in rat brain tissue, taking into account individual resistance to hypoxia and the effects of correction with L-arginine and the nitric oxide synthase inhibitor L-NNA. Specifically, the study aims to a) evaluate the effects of lead nitrate on both early and late stages of lipid peroxidation to determine the effect of lead nitrate toxicity on lipid peroxidation in rat brain tissue; b) investigate the impact of lead exposure on oxidative protein modification in brain tissue and identify key proteins affected by these modifications; c) analyse changes in antioxidant enzyme activity and energy balance enzymes in response to lead nitrate exposure to assess the impact of toxicity on neuronal defence mechanisms and energy metabolism; d) study the acetylcholine-cholinesterase system in rat brain tissue and assess the influence of individual resistance to hypoxia and correction with L-arginine and L-NNA on these relationships.
Materials and Methods
Experimental Animals
The experiments were conducted in accordance with the guidelines of the Council of the European Union, current legislation in Poland and Ukraine, and the recommendations of the Ethics Committee. They were approved by the Ethics Committee of the T.H. Shevchenko National University “Chernihiv Colehium” (28/09/2020). The experiment was conducted in accordance with both Directive 2010/63/EU on the protection of animals used for scientific purposes and the Polish Act of 15 January 2015 on the protection of animals used for scientific or educational purposes (Journal of Laws of 26 February 2015, item 266). All manipulations with the animals were carried out in accordance with the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (Strasbourg, 2006), and every effort was made to minimise animal distress and suffering. Male white Wistar rats (180-220 g) were used in the study. The rats were maintained at a constant temperature of 23 ± 2oC with a 12:12 h light/dark cycle and 40-50% relative humidity. The animals (n = 6 per group) had free access to food and water throughout the experiments.
Determination of Resistance to Hypobaric Hypoxia
The animals were placed in a ventilated pressurised chamber at an ‘altitude’ of 11, 000 m [46–48] once after 15 days of adaptive feeding. A stopwatch was used to record the time to the first sign of the characteristic hyperventilation response (‘gasp time’). Different measures of the gasp time are used to separate animals according to their resistance to hypoxia. However, the gasp time of tolerant animals should be at least three times higher than that of susceptible animals [46–50]. The animals were divided into three groups based on their panting time: ‘low resistance’ (< 80 s), ‘normal’ (80-240 s) and ‘high resistance’ (> 240 s). The ‘normal’ group was excluded from further experiments. Two groups of rats were used for further experiments: rats with low resistance to hypoxia and rats with high resistance to hypoxia [46–50].
Experimental Design
One month after the hypoxia resistance test, the rats were randomly divided into six groups. Group I with low resistance (n = 6) and high resistance to hypoxia (n = 6) served as a control and received daily injections of sterile normal saline for 30 days. Group II (Pb group) with low resistance (n = 6) and high resistance to hypoxia (n = 6) received 3.6 mg lead nitrate/kg b.w. daily by oral gavage. Group III (L-arginine and Pb group) with low resistance (n = 6) and high resistance to hypoxia (n = 6) also received 3.6 mg lead nitrate/kg b.w. daily for 30 days; during this time, the animals were intraperitoneally (i.p.) injected with L-arginine at a dose of 600 mg/kg b.w. before the 30-min lead nitrate treatment. Group IV (L-NNA and Pb group) was treated as group III except that it received an L-NNA injection (35 mg/kg b.w.) before 30 minutes of lead nitrate treatment. Group V (Pb group and L-arginine) with low resistance (n = 6) and high resistance to hypoxia (n = 6) also received 3.6 mg lead nitrate/kg b.w. daily for 30 days, during which the animals were injected intraperitoneally (i.p.) with L-arginine at a dose of 600 mg/kg b.w. at 30 minutes after the lead nitrate treatment. Group VI (Pb group and L-NNA) was treated as group V except that it received an L-NNA injection (35 mg/kg b.w.) at 30 minutes after the lead nitrate treatment. In summary, the experimental protocol of this study was based on 12 groups. The doses of L-arginine and L-NNA were chosen on the basis of our previous studies [51–53]. All rats were euthanised by intraperitoneal injection of a lethal dose of sodium pentobarbital (Morbital, Biowet, Pulawy; 200 mg/kg b.w.) on day 31 of the experiment.
The brain was rapidly removed and homogenised at 4oC in 6 volumes of 0.1 M Tris-HCl buffer (pH 7.4) using a glass homogeniser with a Teflon pestle attached to a motor drive, and the volume was adjusted to give a 10% w/v homogenate. The homogenate was centrifuged and the supernatant was used for the estimation of various biochemical parameters. The protein content in the brain tissue homogenate was estimated using bovine serum albumin as a standard [54].
Biochemical Estimations and Assays
Statistical Analysis
The statistical analysis was performed using the STATISTICA 13.3 software package (TIBCO Inc., USA), which allows basic statistical evaluations, including significance of regression slopes, analysis of variance to assess group differences, and distribution tests. Levene’s test was used to test for homogeneity of variance, while the Kolmogorov-Smirnov test was used to assess normality of the data. The results are presented as mean ± standard deviation (S.D.). Significant differences between means were determined by a multiple range test at a significance level of P < 0.05. Logarithmic transformations were used for data that were not normally distributed. Differences between the groups were assessed using Student’s t-tests with 95% confidence intervals (α = 0.05). Parametric correlations, including correlation coefficients (r), regression equations, and their significance (P), were examined using Pearson’s analysis. ANOVA was used to estimate the mean concentrations of biomarkers and enzyme activity in brain tissue. Statistically significant relation-ships across all values were identified by applying significance tests to the effects of varying resistance to hypoxia.
Results
Lipid Peroxidation Processes
Changes in the levels of DC (A) and TBARS (B) are shown in Fig. 3. The per os administration of lead nitrate to the rats was associated with a statistically significant increase in the levels of both DC and TBARS in the brain tissue, compared with untreated rats. These changes in DC were significant in the group of animals with low (LR) and high (HR) resistance to hypoxia and exhibited the same level of dependence. For TBARS, these changes were statistically significantly higher in the HR individuals compared to the LR animals in both the untreated control and the lead-exposed groups, compared to the control values. Thus, it is important to investigate individual factors of physiological reactivity under the influence of lead, and the initial and terminal steps of the lipoperoxidation process are differentially involved in these processes in brain tissue.
As L-arginine plays a key role in maintaining brain health by promoting neurotransmission, blood flow, and protection against oxidative stress through the production of nitric oxide. Since L-NNA as an inhibitor of nitric oxide synthase reduces NO production, which can lead to disturbances in brain function, this approach was used to assess the effects of lead nitrate, taking into account the different physiological reactivity of the body. The level of DС in brain tissue after administration of L-arginine prior to lead nitrate intoxication was significantly increased only in the group of animals with HR. In contrast, the administration of the nitric oxide synthase inhibitor L-NNA in these conditions was associated with a significant increase in the level of DС in both LR and HR rats, compared with the data from the lead nitrate-exposed group (Fig. 3A). It should be noted that the difference between the effects of L-NNA and exposure to lead nitrate in the rats from both LR and HR groups was also statistically expressed, i.e. the effects of L-NNA in the group of HR animals were manifested in a more pronounced decrease in the level of lipoperoxidation processes at the initial stages of this process than in the group with LR. However, the levels remained above those of the group exposed to lead nitrate. This tendency was also maintained in experimental conditions in which L-arginine was administered after the exposure to lead nitrate, i.e. both L-arginine and L-NNA increased the values of the DC level in the rats, compared with the data obtained in the group exposed to lead nitrate alone.
The effects of L-arginine under the lead nitrate exposure were accompanied by a statistically significant decrease in TBARS levels in the LR group; we observed the same effects in the HR animals when L-NNA was administered (Fig. 3B). It should be noted that this difference in the effects of L-arginine and L-NNA under the lead nitrate exposure between the groups was statistically significant. This difference in the effects of the enhancement (when L-arginine was administered) and reduction (when L-NNA, a nitric oxide synthase inhibitor, was administered) of the role of the nitric oxide system in the brain tissue at the level of the end products of the lipid peroxidation process was maintained in these studies when the preparations were administered after the exposure to lead nitrate.
Therefore, the individual factors of physiological reactivity are crucial in investigations of the effects of lead nitrate exposure. Our research suggests that the initial and final products of the lipoperoxidation process are differently involved in lead-induced changes in brain tissue. The analysis of the effects of L-arginine on the lead nitrate exposed animals revealed a statistically significant reduction in TBARS levels in the brain tissue of the LR rats, and the administration of L-NNA to the HR animals produced comparable effects. Importantly, the differences observed in the effects of L-arginine and L-NNA on the lead nitrate exposed rats were statistically significant between the LR and HR groups.
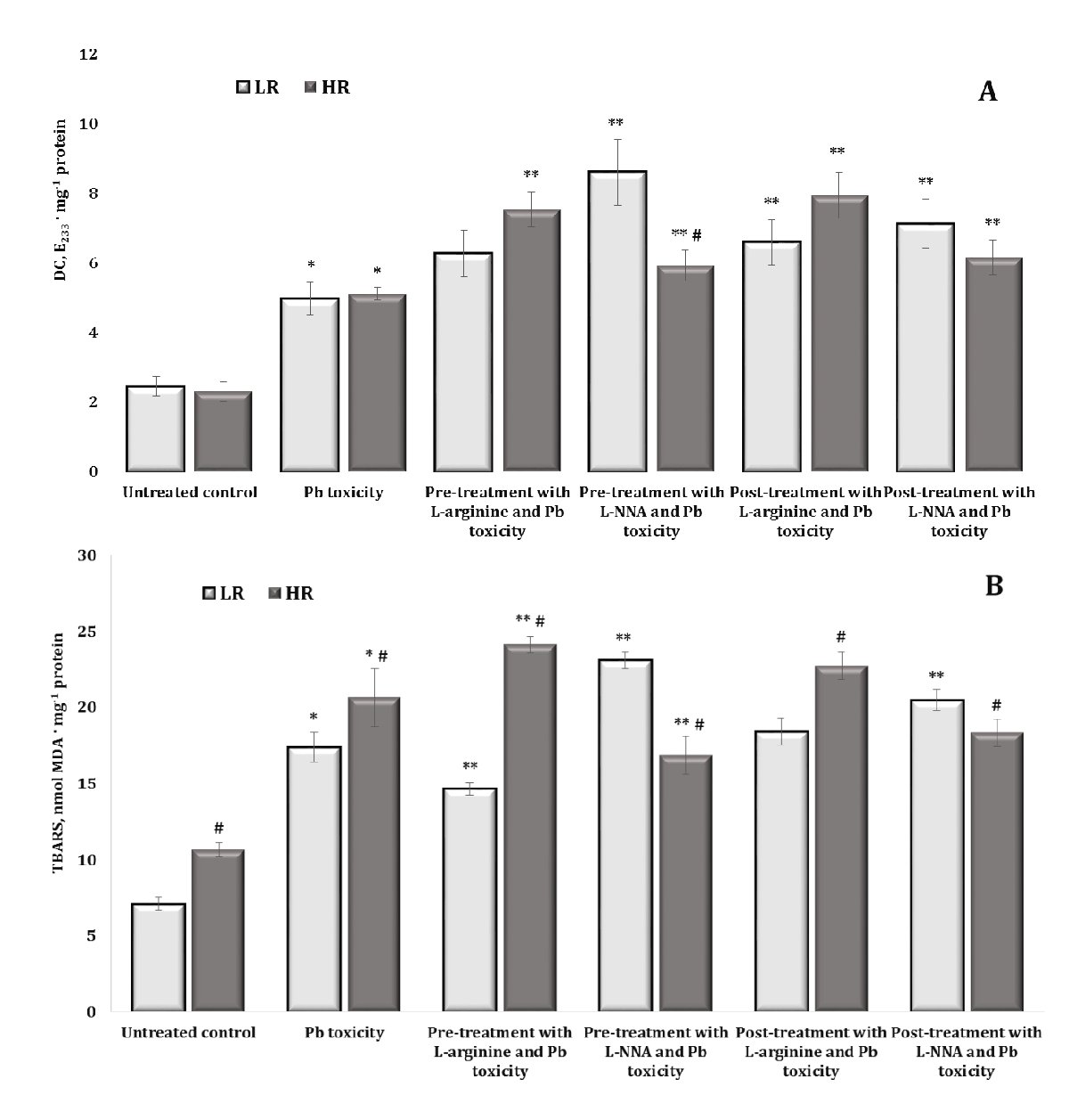
Fig. 3: Levels of diene conjugates (DC, E233 ∙ mg-1 protein, A) and TBARS (nmol MDA ∙ mg-1 protein, B) in the brain tissue of rats with low (LR) and high (HR) hypoxia resistance exposed to lead nitrate for 30 days and treated with L-arginine (600 mg/kg, 30 min) or L-NNA (35 mg/kg, 30 min) before and after the exposure to lead nitrate. Data are expressed as mean ± S.D. (n = 6). * Significant differences between the Pb group and the untreated control (p < 0.05); ** Significant differences between the Pb group and the L-arginine + Pb group, the L-NNA + Pb group, the Pb + L-arginine group, and the Pb + L-NNA group (p < 0.05); # Significant differences between the LR and HR groups in the same conditions (p < 0.05).
Oxidatively Modified Proteins
Oxidative modification of proteins is a critical mechanism in lead-induced neurotoxicity, causing significant changes in the structure and function of neuronal proteins. The results of lead-induced neurotoxicity for carbonyl derivatives of protein oxidation shown in Fig. 4 revealed an increase in the levels of aldehyde and ketone groups on amino acid residues and the formation of neutral dinitrophenyl hydrazines. In fact, the data shown in Fig. 4 and Fig. 5 represent aldehyde and ketone groups on amino acid residues forming neutral aliphatic aldehyde dinitrophenylhydrazones (OMP ADn, Fig. 4A), neutral aliphatic ketone dinitrophenylhydrazones (OMP KDn, Fig. 4B), alkaline aliphatic aldehyde dinitrophenylhydrazones (OMP ADa, Fig. 5A), and alkaline aliphatic ketone dinitrophenylhydrazones (OMP KDa, Fig. 5A).
In our study, the oral administration of lead nitrate to both low and high hypoxia-resistant animals resulted in a statistically significant increase in oxidatively modified protein (OMP) levels in their brain tissue, compared to the controls. This suggests that lead nitrate induces oxidative stress in the brain, as evidenced by the increased levels of oxidatively modified proteins. We showed differential effects based on resistance to hypoxia, namely the increase in the OMP levels was significantly higher in the low hypoxia-resistant rats than in the high hypoxia-resistant rats. Specifically, in the low resistance group of rats, the levels of oxidatively modified proteins increased by 136.7% (p < 0.05) for OMP ADn, 109.7% (p < 0.05) for OMP KDn, 93.9% (p < 0.05) for OMP Ada, and 85.1% (p < 0.05) for OMP KDa. In the highly resistant group, the increases were less pronounced but still statistically significant: 60% (p < 0.05) for OMP ADn, 80.3% (p < 0.05) for OMP KDn, 83.7% (p < 0.05) for OMP ADa, and 75.4% (p < 0.05) for OMP KDa. We therefore investigated the role of hypoxia resistance on the oxidative response of proteins in brain tissue. The results suggest that although both low and high hypoxia-resistant animals experience oxidative stress due to lead nitrate exposure, the extent of oxidative protein modifications is greater in animals with lower hypoxia resistance. This suggests potential differential sensitivity to oxidative damage based on the hypoxia resistance of the animal.
The effects of administering L-arginine and L-NNA to the animals with low and high hypoxia resistance exposed to lead nitrate showed statistically significant differences in the levels of oxidatively modified proteins in brain tissue, compared to the levels in the animals exposed to lead nitrate alone. In other words, the administration of L-arginine and L-NNA had statistically significant opposite effects and depended on the rats’ initial resistance to hypoxia. Indeed, no statistically significant differences were observed in the levels of OMP ADn (Fig. 4A) after L-arginine administration, but the differences in the effects of this precursor of nitric oxide synthesis between the two LR and HR groups were statistically significant in relation to the hypoxia resistance factor. This trend was observed both when L-arginine was administered to the rats before the exposure to lead nitrate and in the group of rats after the exposure to lead nitrate. In the case of the OMP ADn values in the group of highly resistant rats, we obtained a pronounced dependence of the OMP reduction, compared to the data obtained with the lead nitrate exposure, where exactly L-NNA caused a reduction of the OMP ADn parameter when administered both before and after the lead nitrate exposure. Thus, the effects of the modulation of the NO system are important during lead nitrate exposure, whereas the effects of the NO synthase inhibitor L-NNA are essential in the pre-treatment of oxidative-induced disorders in groups of individuals with high individual physiological status.
The effects of the L-NNA administration led to a significant decrease in the levels of OMP KDn (Fig. 4B) and OMP KDa (Fig. 5A) only in the group of high resistance rats in both types of lead exposure (before and after); this dependence was maintained for OMP ADa levels when L-NNA was administered to the rats only after the lead nitrate exposure. In contrast, in the group of low resistance rats, we obtained a significant decrease in OMP KDa levels when L-arginine was administered both before and after the lead nitrate exposure (Fig. 5B). Thus, in the rats with low resistance to hypoxia, the administration of L-arginine before and after the exposure to lead nitrate resulted in a significant decrease in OMP KDa levels, suggesting that L-arginine may help to mitigate some of the oxidative damage induced by lead nitrate in this group. Conversely, in the high hypoxia-resistant rats, L-NNA, a nitric oxide synthase inhibitor, led to a significant reduction in OMP KDn and OMP KDa levels in both types of lead exposure (pre- and post-exposure). Furthermore, such a reduction was also observed for OMP ADa (Fig. 5A) when L-NNA was administered only after the lead nitrate exposure, indicating that the inhibition of nitric oxide synthesis has a pronounced effect on oxidative protein modification, specifically in highly resistant rats.
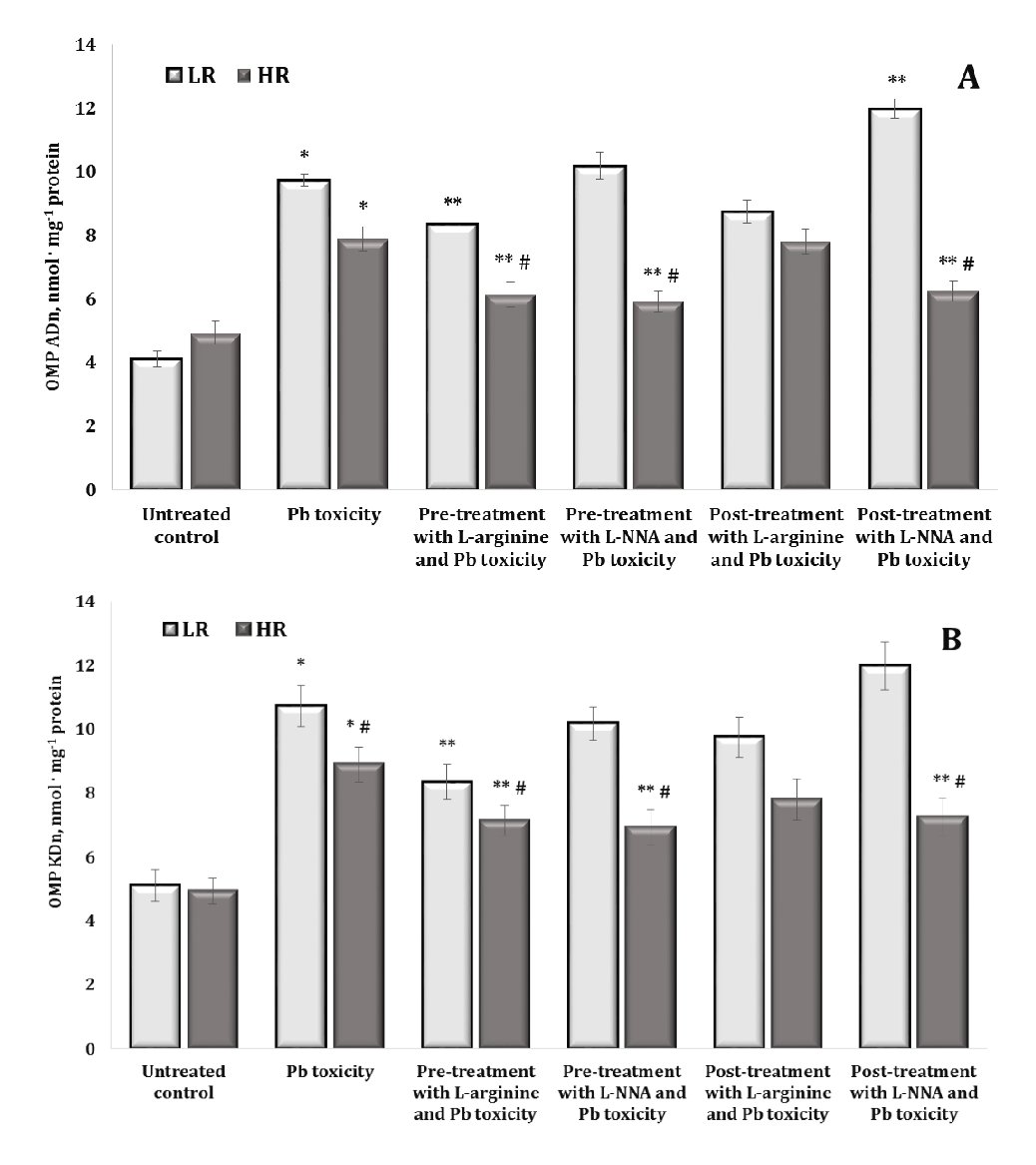
Fig. 4: Levels of neutral aliphatic aldehyde dinitrophenyl hydrazones (OMP ADn, A) and neutral aliphatic ketone dinitrophenyl hydrazones (OMP KDn, B) in the brain tissue of rats with low (LR) and high (HR) hypoxia resistance exposed to lead nitrate for 30 days and treated with L-arginine (600 mg/kg, 30 min) or L-NNA (35 mg/kg, 30 min) before and after the exposure to lead nitrate. Data are expressed as mean ± S.D. (n = 6). * Significant differences between the Pb group and the untreated control (p < 0.05); ** Significant differences between the Pb group and the L-arginine + Pb group, the L-NNA + Pb group, the Pb + L-arginine group, and the Pb + L-NNA group (p < 0.05); # Significant differences between the LR and HR groups in the same conditions (p < 0.05).
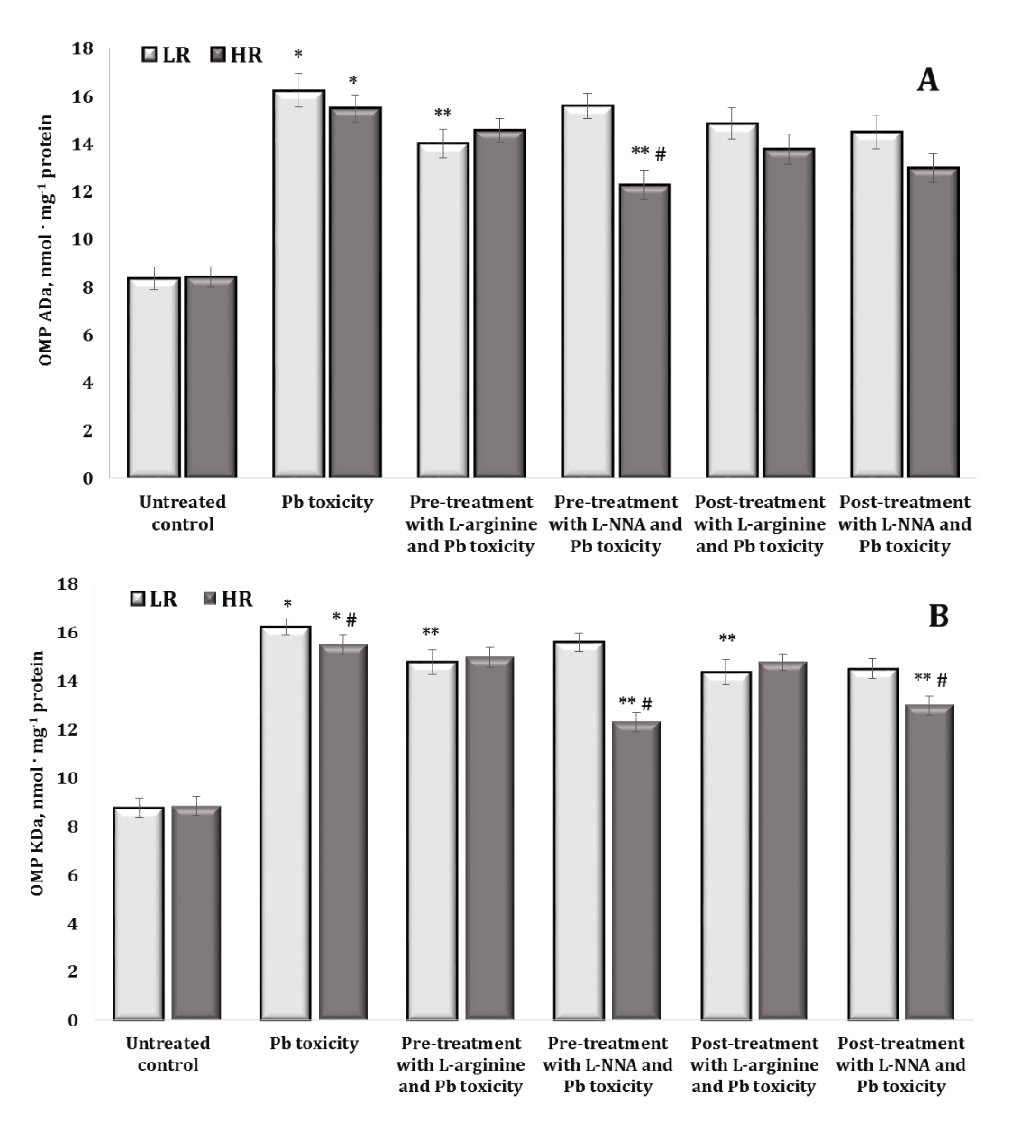
Fig. 5: Levels of alkaline aliphatic aldehyde dinitrophenyl hydrazones (OMP ADa, A) and alkaline aliphatic ketone dinitrophenylhydrazones (OMP KDa, B) in the brain tissue of rats with low (LR) and high (HR) hypoxia resistance exposed to lead nitrate for 30 days and treated with L-arginine (600 mg/kg, 30 min) or L-NNA (35 mg/kg, 30 min) before and after the exposure to lead nitrate. Data are expressed as mean ± S.D. (n = 6). * Significant differences between the Pb group and the untreated control (p < 0.05); ** Significant differences between the Pb group and the L-arginine + Pb group, the L-NNA + Pb group, the Pb + L-arginine group, and the Pb + L-NNA group (p < 0.05); # Significant differences between the LR and HR groups in the same conditions (p < 0.05).
Activity of Antioxidant Enzymes
The assessment of antioxidant enzyme activity [superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), and glutathione reductase (GR)] was the next stage of our research into the relationship between lead nitrate exposure and modulation of the nitric oxide system in rat brain tissue. This step also focused on understanding the influence of individual physiological reactivity parameters on this relationship. By looking at enzyme activities, we aimed to gain a deeper insight into the effect of nitric oxide pathways on oxidative stress induced by lead nitrate exposure and the potential impact of individual variations in physiological responses on these processes. The results are shown in Figures 6 and 7.
Superoxide dismutase (SOD), which converts superoxide radicals to oxygen and hydrogen peroxide, showed the highest levels in the highly resistant rats in the control group, compared to the low resistant rats (Fig. 6A). The effect of the lead nitrate exposure in the highly resistant animals was accompanied by a significant decrease in SOD activity. We observed a similar decrease in the activity of this enzyme in the group of low resistant animals when L-NNA was administered (both before and after the lead nitrate exposure). The effects of L-arginine in both the LR and HR rats were accompanied by an increase in SOD activity when this amino acid was administered after the lead nitrate exposure.
The effects of the lead nitrate exposure on the activity of CAT, which reduces hydrogen peroxide (H₂O₂) to water and oxygen, thus preventing the accumulation of this potentially harmful compound, are shown in Fig. 6B. Our results showed that the exposure to lead nitrate induced an increase in CAT activity in the two groups of LR and HR rats. We noted that, in these experimental conditions, the effect of both L-arginine and the inhibitor L-NNA caused a statistically significant increase in CAT activity under the lead nitrate exposure, but these dependencies were maintained at the level of individual physiological reactivity associated with different resistance to hypoxia.
Thus, CAT plays a critical role in neutralising hydrogen peroxide in brain tissue during lead nitrate exposure and its activity is essential for protecting cells from oxidative stress. L-arginine, as a precursor of nitric oxide, and its interactions with hydrogen peroxide may affect the levels and function of CAT and depend on individual resistance to hypoxia. It is possible that increased production of hydrogen peroxide stimulates CAT activity and, conversely, reactions between NO and H₂O₂ may lead to the formation of peroxynitrite (ONOO-), which may further suppress cellular defence mechanisms and affect CAT function.
The study of glutathione metabolism-dependent enzymes, such as glutathione reductase (GR) and glutathione peroxidase (GPx), in lead nitrate exposure is crucial because these enzymes play a fundamental role in neutralising free radicals and protecting cells from oxidative stress. The data are shown in Fig. 7A,B. Examination of GR activity in our study after the exposure to lead nitrate and treatment with L-arginine or L-NNA showed that these relationships were not observed in the group of animals with low resistance. In the group of highly resistant rats, these changes showed increased GR activity induced by the lead nitrate exposure, compared to the data obtained in the untreated group, and were statistically reduced in all combinations of our experiment with L-arginine and L-NNA. It should be noted that we observed significant differences in the effects of the lead nitrate exposure and L-NNA treatment between the groups of high and low resistant animals (Fig. 7A).
The examination of GPx activity after the lead nitrate exposure revealed a statistically significant decrease in the activity of this enzyme in both groups of LR and HR rats, compared to the untreated groups (Fig. 7B). The administration of both L-arginine and L-NNA resulted in a statistically significant increase in the activity of this enzyme in both groups of LR and HR rats, both when L-arginine and L-NNA were administered before and after the exposure to lead nitrate. It should be noted that when L-NNA was administered both before and after the lead nitrate exposure, significantly higher activity was observed than in the case of the same analysed effects of L-arginine. Thus, the effect of the nitric oxide synthase inhibitor L-NNA during the lead nitrate exposure induced a more pronounced increase in GPx activity in the brain tissue than the effect of the amino acid L-arginine and was slightly dependent on individual physiological reactivity (Fig. 7B).
The next stage of our study was to examine the total antioxidant status (TAS) of brain tissue, which includes both enzymatic and non-enzymatic systems. The data are presented in Fig. 8.
It should be noted that higher TAS values were observed in the untreated control group of highly resistant rats, compared to the LR rats (by 44.8%, p < 0.05). Thus, the higher capacity of the antioxidant defence of functional brain systems shown in the control group of highly resistant animals, compared to the data obtained in the low resistant animals, is important for understanding the initial physiological differences in response to the influence of factors of different origin. The effects of the lead nitrate exposure were associated with a statistically significant decrease in the TAS levels, and these changes were more pronounced in the group of highly resistant rats. The beneficial effects of L-arginine were associated with a significant increase in the TAS levels in both groups of LR and HR rats during the lead nitrate exposure both before and after the L-arginine administration, and this difference in the effects of the precursor of nitric oxide synthesis was also significant between the groups of LR and HR animals. In the brain tissue, the effects of L-NNA and lead nitrate exposure offset the effects of L-arginine in the LR rats, and only in the HR group when L-NNA was administered after the lead nitrate exposure were the levels higher than in the lead nitrate exposure group.
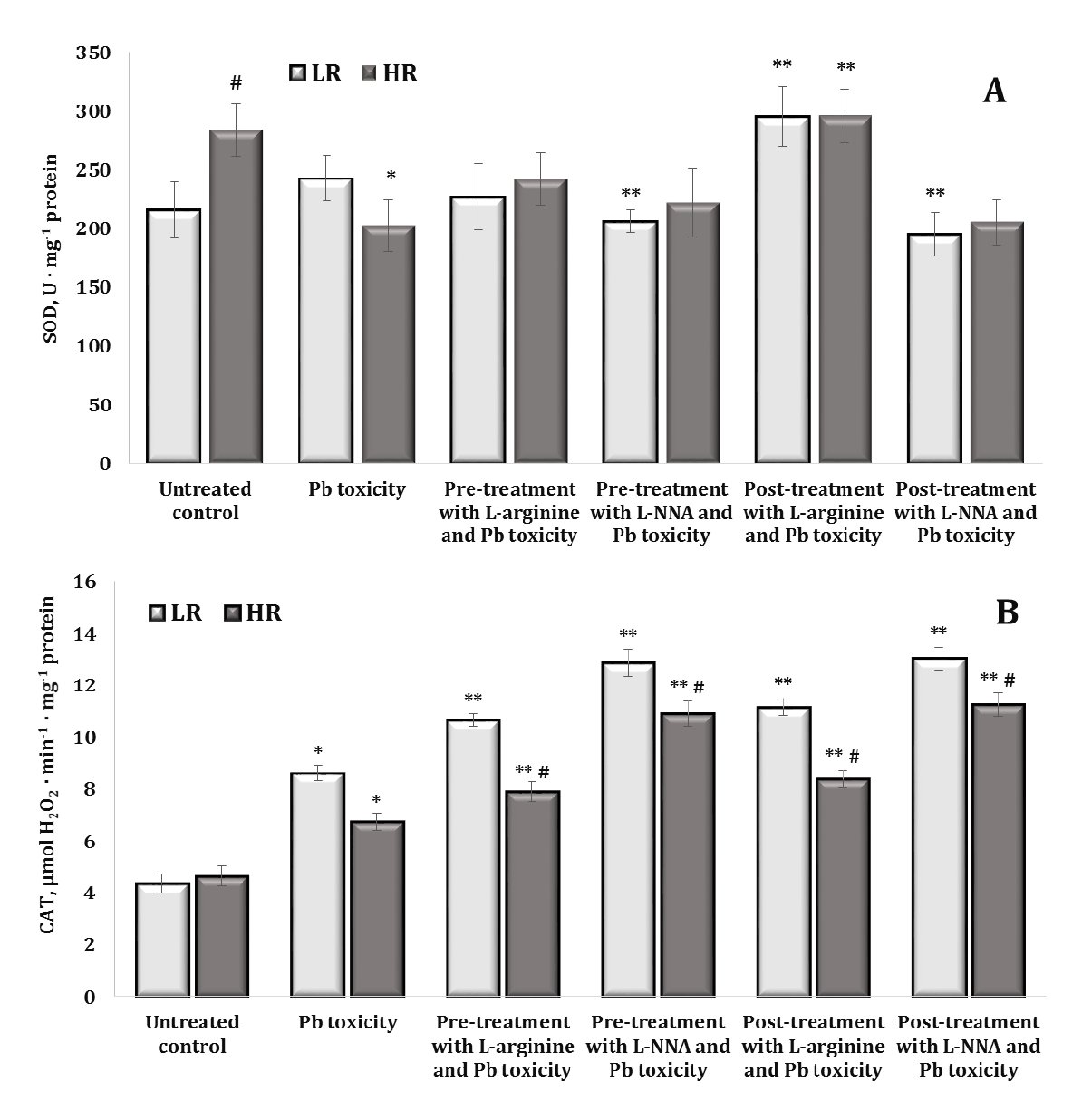
Fig. 6: Activity of superoxide dismutase (SOD, U ∙ mg-1 protein, A) and catalase (μmol H2O2 · min-1 · mg-1 protein, B) in the brain tissue of rats with low (LR) and high (HR) hypoxia resistance exposed to lead nitrate for 30 days and treated with L-arginine (600 mg/kg, 30 min) or L-NNA (35 mg/kg, 30 min) before and after the exposure to lead nitrate. Data are expressed as mean ± S.D. (n = 6). * Significant differences between the Pb group and the untreated control (p < 0.05); ** Significant differences between the Pb group and the L-arginine + Pb group, the L-NNA + Pb group, the Pb + L-arginine group, and the Pb + L-NNA group (p < 0.05); # Significant differences between the LR and HR groups in the same conditions (p < 0.05).
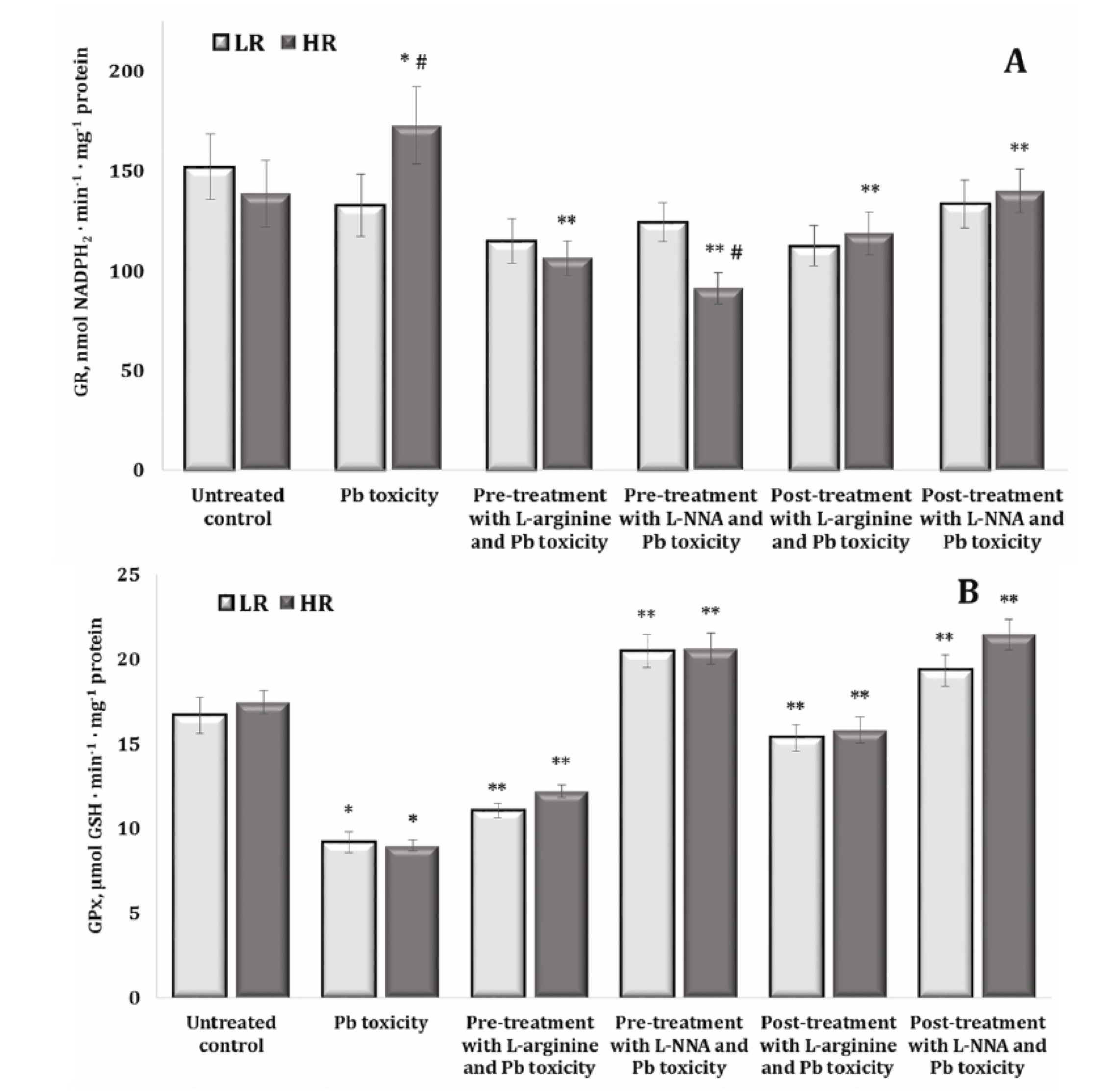
Fig. 7: Activity of glutathione reductase (GR, U · mg-1 protein, A), and glutathione peroxidase (GPx, U · mg-1 protein, B) in the brain tissue of rats with low (LR) and high (HR) hypoxia resistance exposed to lead nitrate for 30 days and treated with L-arginine (600 mg/kg, 30 min) or L-NNA (35 mg/kg, 30 min) before and after the exposure to lead nitrate. Data are expressed as mean ± S.D. (n = 6). * Significant differences between the Pb group and the untreated control (p < 0.05); ** Significant differences between the Pb group and the L-arginine + Pb group, the L-NNA + Pb group, the Pb + L-arginine group, and the Pb + L-NNA group (p < 0.05); # Significant differences between the LR and HR groups in the same conditions (p < 0.05).
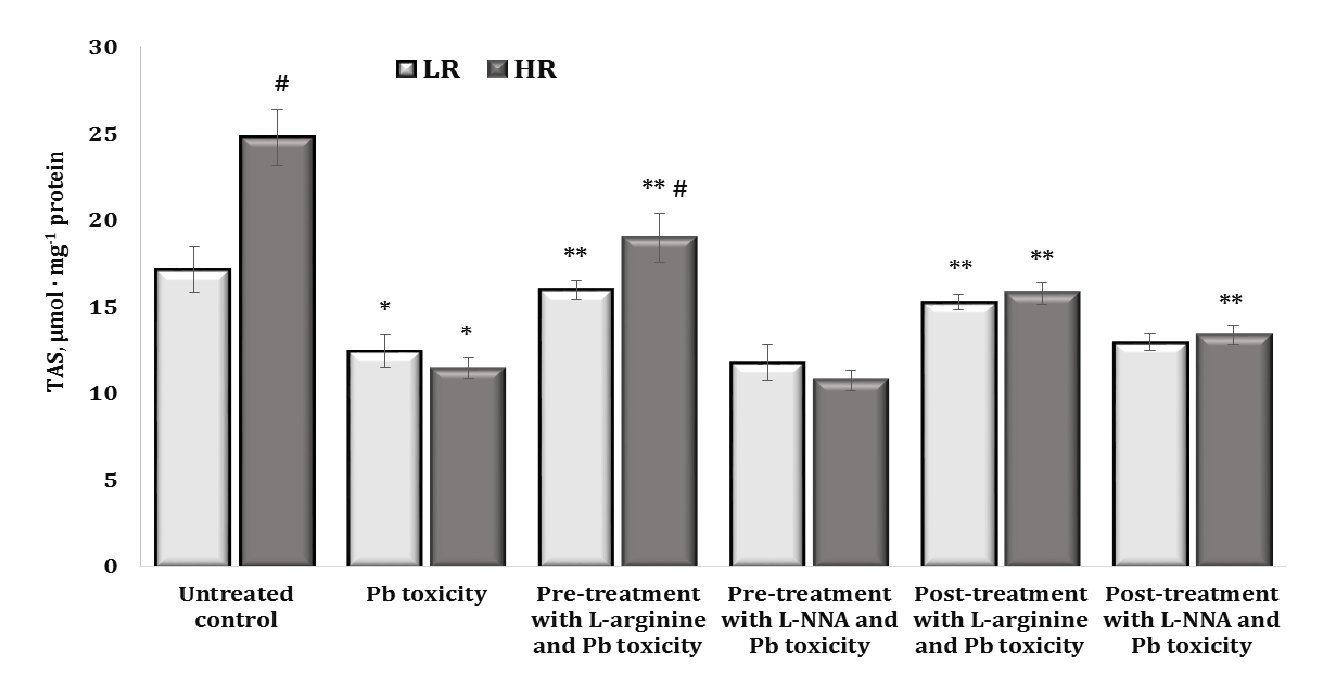
Fig. 8: Total antioxidant status (TAS, µmol · mg-1 protein) in the brain tissue of rats with low (LR) and high (HR) hypoxia resistance exposed to lead nitrate for 30 days and treated with L-arginine (600 mg/kg, 30 min) or L-NNA (35 mg/kg, 30 min) before and after the exposure to lead nitrate. Data are expressed as mean ± S.D. (n = 6). * Significant differences between the Pb group and the untreated control (p < 0.05); ** Significant differences between the Pb group and the L-arginine + Pb group, the L-NNA + Pb group, the Pb + L-arginine group, and the Pb + L-NNA group (p < 0.05); # Significant differences between the LR and HR groups in the same conditions (p < 0.05).
Biochemical Pathway Analysis
The pathways involving alanine and aspartate aminotransferases, succinate dehydrogenase, and α-ketoglutarate dehydrogenase are interconnected through such key intermediates as glutamate, α-ketoglutarate, succinate, and oxaloacetate, which integrate amino acid metabolism with the citric acid cycle (Krebs cycle); hence, the analysis of their interdependencies was the next step of our study. These changes are shown in Table 1.
In our studies, we observed a significant increase in ALT and AST activity in brain tissue under the influence of lead nitrate. These increases in transamination enzyme activity were more pronounced under the influence of L-arginine administered with lead nitrate and were reversed by the administration of the nitric oxide synthase inhibitor, L-NNA, compared with data obtained in the untreated control groups. These changes were also statistically significant between the HR and LR rats when L-arginine and lead nitrate were administered. It is important to note that the de Ritis coefficient (the ratio between the activity of AST and ALT) in our studies was dependent on the type of nitric oxide modulators used (L-arginine and L-NNA). Elevated values of this coefficient often indicate an unfavourable prognosis in the course of pathological processes. Thus, in our studies, the de Ritis coefficient was reduced in both groups of LR and HR animals by the administration of L-arginine, and the administration of the nitric oxide synthase inhibitor L-NNA reduced these dependencies (Table 1).
The analysis of pathway integration processes by SDH and KGDH is important because SDH catalyses the conversion of succinate to fumarate and the FADH₂ formed in this reaction transfers electrons to the respiratory chain, which is crucial for ATP production. SDH is the only Krebs cycle enzyme that is also part of the respiratory chain, highlighting its key role in the integration of these two processes, and the succinate formed by KGDH is the substrate for SDH, linking the Krebs cycle to energy production by the mitochondrial respiratory chain. In our studies, we observed multidirectional effects of lead nitrate in brain tissue, and these dependencies were determined by the resistance of the animals to hypoxia. The activity of SDH, which catalyses the conversion of succinate to fumarate and the resulting FADH₂, which transfers electrons to the respiratory chain important for ATP production, was increased in the low resistance animals and statistically decreased in the high resistance animals (Table 1). The administration of L-arginine before the lead nitrate exposure decreased SDH activity in the LR group and increased the SDH activity in the HR group, compared with data from these groups exposed to lead nitrate alone. When L-NNA was administered after the lead nitrate exposure, the SDH activity was statistically increased in both groups of LR and HR animals. KGDH activity was statistically increased in both groups of LR and HR animals when L-arginine was administered before the lead nitrate exposure, compared to data obtained from the lead nitrate exposure group. Similar effects of increased KGDH activity in the brain tissue were also exerted by the L-NNA administration (Table 1).
Thus, our study shows that lead nitrate exposure significantly increases brain ALT and AST activity, with L-arginine exacerbating and L-NNA reversing this effect. The de Ritis coefficient, a marker of pathological prognosis, was reduced by L-arginine and further reduced by L-NNA. The lead nitrate exposure also affected the activities of SDH and KGDH, which are crucial for cellular energy production and hypoxia resistance, with L-arginine altering SDH activity, depending on the level of animal resistance to hypoxia and L-NNA increasing both SDH and KGDH activities (Table 1). These findings highlight the potential of modulators of the nitric oxide system to counteract lead-induced biochemical dysfunction and show that this potential is dependent on the initial physiological characteristics of the organisms.
Acetylcholine-Cholinesterase System
In our studies, we observed a statistically significant difference in the levels of acetylcholine (ACh) and the activity of its hydrolysing enzyme acetylcholinesterase (AChE) in the brain tissue of high and low resistant rats, with the highest levels of substrate and enzyme activity in highly resistant individuals. These relationships are illustrated in Fig. 9.
Thus, the dependencies on the initial mechanisms of resistance to hypoxia determine the underlying mechanisms of metabolic processes. The oral exposure to lead nitrate significantly increased ACh levels and significantly decreased AChE activity, compared to the untreated controls. However, these effects were statistically dependent on the level of hypoxia resistance. The administration of L-arginine prior to the exposure to lead nitrate was associated with a significant decrease in ACh levels with an increase in AChE activity in the group of highly resistant rats, and a significant increase in AChE activity alone was demonstrated in the group of LR rats. The effects of L-NNA and the lead nitrate exposure caused a decrease in ACh levels and maintenance of high AChE activity in the LR rats and were accompanied by high AChE activity in the highly resistant rats, compared to those obtained in the lead nitrate exposure group. In the case of the effects of L-arginine and L-NNA, we observed similar effects of ACh-AChE dependence when L-arginine and L-NNA were administered after the oral lead nitrate exposure (Fig. 9A,B).
Thus, our results suggest that therapeutic approaches targeting modulation of nitric oxide pathways, assessed by lipoperoxidation, antioxidant enzyme activity, biochemical pathways involving alanine and aspartate aminotransferases, succinate dehydrogenase, and α-ketoglutarate dehydrogenase, and the acetylcholine-cholinesterase system, are critical in counteracting oxidative damage from lead nitrate exposure, especially when considering individual hypoxia resistance. For those with low hypoxia resistance, L-arginine, which increases nitric oxide production, can provide significant protection by reducing oxidative damage in brain tissue. This protection is evidenced by reduced levels of oxidatively modified proteins and lipoperoxidation as well as improved antioxidant enzyme activity. On the other hand, the use of L-NNA, a nitric oxide synthase inhibitor, appears to be more effective in reducing oxidative modifications in individuals with high hypoxia resistance. This is reflected in a significant reduction in oxidatively modified proteins and lipoperoxidation as well as potential effects on the acetylcholine-cholinesterase system, which could influence neurotransmitter dynamics and oxidative stress responses. Therefore, effective treatment strategies should be personalised based on hypoxia resistance and include strategies to modulate nitric oxide levels, manage lipoperoxidation, enhance antioxidant defences and regulate the acetylcholine-cholinesterase system to combat lead-induced oxidative stress.
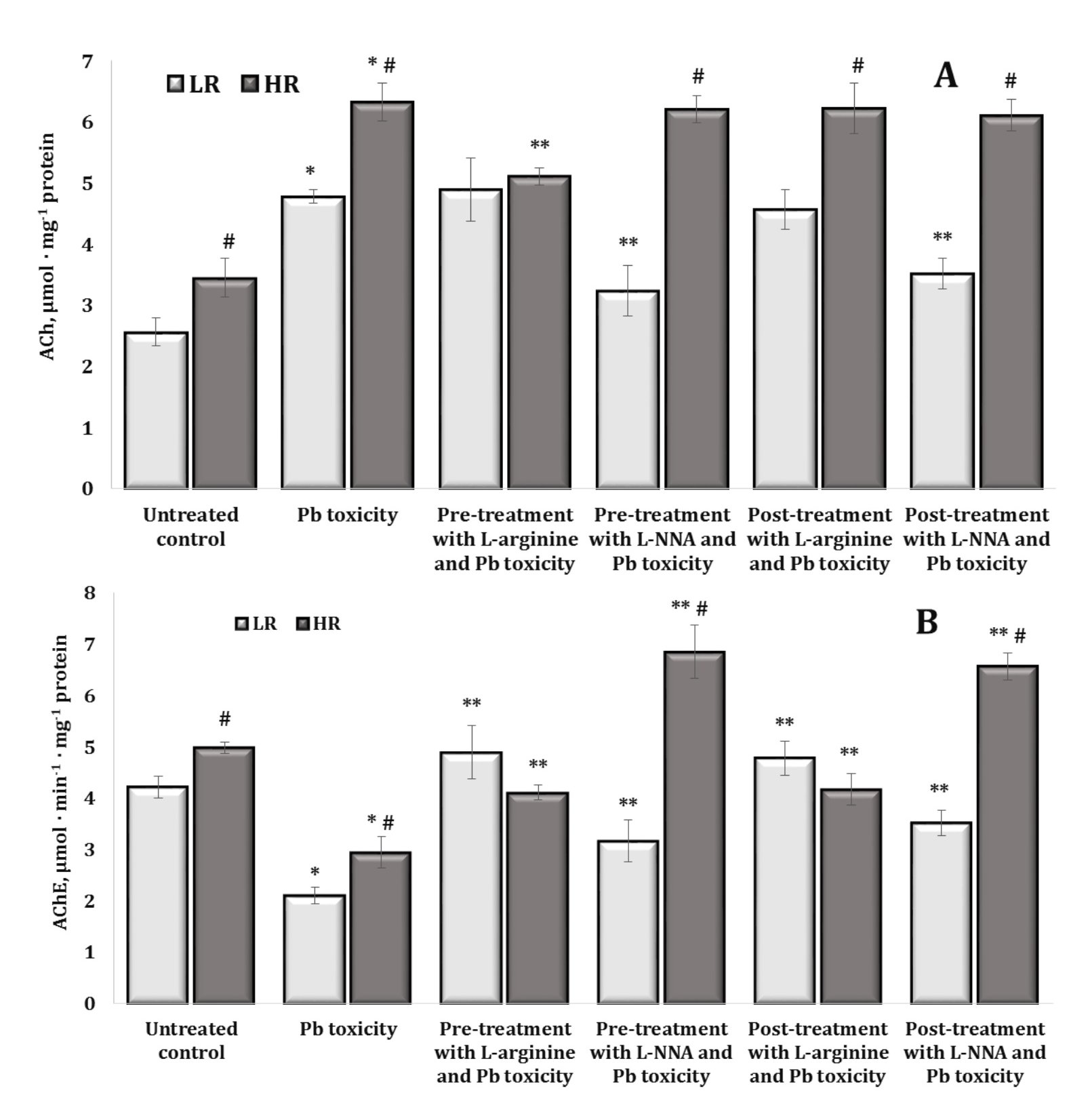
Fig. 9: Acetylcholine content (ACh, µmol · mg-1 protein, A) and acetylcholine esterase activity (AChE, µmol · min-1 · mg-1 protein, B) in the brain tissue of rats with low (LR) and high (HR) hypoxia resistance exposed to lead nitrate for 30 days and treated with L-arginine (600 mg/kg, 30 min) or L-NNA (35 mg/kg, 30 min) before and after the exposure to lead nitrate. Data are expressed as mean ± S.D. (n = 6). * Significant differences between the Pb group and the untreated control (p < 0.05); ** Significant differences between the Pb group and the L-arginine + Pb group, the L-NNA + Pb group, the Pb + L-arginine group, and the Pb + L-NNA group (p < 0.05); # Significant differences between the LR and HR groups in the same conditions (p < 0.05).
Discussion
In this study, we investigated the effects of oral exposure to lead nitrate on a variety of oxidative and energetic processes in rat brain tissue. We analysed biomarkers of the early and late stages of lipid peroxidation to assess the effect of lead nitrate toxicity on lipid damage. We also examined the levels of carbonyl derivatives to identify oxidative modifications of proteins altered by lead nitrate and assessed changes in antioxidant enzyme activity and biomarkers of energy processes to understand the effects of oral lead nitrate exposure on rat brain tissue metabolism. We also examined the relationship between the acetylcholine and cholinesterase systems, considering the impact of individual resistance to hypoxia and correction of lead nitrate toxicity by L-arginine and L-NNA on these interactions. We attempted to highlight the most important aspects of the results by detailing the key findings in the following points. This approach allowed the key findings of our study to be systematically presented and discussed, providing a clear and comprehensive understanding of the effect of oral lead nitrate exposure on oxidative and energetic processes in rat brain tissue. By focusing on these critical points, we illustrated the broader implications of our findings and their relevance to the management of lead toxicity and its effects on brain tissue health (Fig. 10).
Fig. 10: NO in modulation of lead neurotoxicity in rats with different levels of hypoxic tolerance.
Firstly, this research is of great importance as it focuses on understanding the complex interactions between lead nitrate exposure and oxidative stress in brain tissue, a topic that remains critically relevant due to the ongoing presence of lead contamination and its detrimental effects on public health [5]. Lead exposure continues to be a major environmental health concern, particularly in areas of historical contamination [73], and understanding its mechanisms of action is essential for the development of effective interventions and protective measures [74]. Importantly, the focus of our study on individual resistance to stress-related factors, such as hypoxia, and the modulation of lead toxicity by L-arginine and L-NNA is particularly timely and relevant. While many studies have investigated the general effects of lead on oxidative stress [75–77], there is a notable gap in understanding the influence of individual variability in nitric oxide signalling on susceptibility to lead toxicity. Investigating this aspect could provide new insights into personalised approaches to mitigate lead-induced damage, which is crucial for improving therapeutic strategies.
The brain barrier is the primary target of systemic lead exposure, with this element entering the brain via several mechanisms that disrupt the neurophysiological functions by which lead infiltrates the brain and explains its neurotoxic mechanisms [78]. These include ion mimicry, where Pb²⁺ replaces essential metal ions, mitochondrial dysfunction, redox imbalance caused by oxidative stress, and neuroinflammation. Given these mechanisms, pharmacological interventions are crucial to mitigate the deleterious effects of lead through the efficacy of chelators, antioxidants, anti-inflammatory drugs, and their combinations as integrated therapeutic strategies [1, 78]. These approaches aim to counteract lead toxicity by enhancing detoxification, neutralising oxidative damage, and reducing inflammation, thus offering potential solutions to protect both developing and adult organisms from lead-induced neurotoxicity [79].
In this context, lead affects NO function in the brain through several additional mechanisms [12, 80]. First, lead induces oxidative stress, leading to the production of reactive oxygen species (ROS), which can react with NO to form peroxynitrite, a highly reactive oxidant that further damages cellular components [12]. This interaction exacerbates oxidative damage and disrupts NO function via synaptic function [81]. Lead also impairs redox homeostasis by reducing the levels of key antioxidants, such as glutathione, which are essential for neutralising ROS. The resulting imbalance in redox status contributes to increased oxidative damage and disrupts normal NO signalling. In addition, Pb affects the expression of genes involved in NO production and its regulatory proteins, thereby altering NO synthesis and function in the brain [82]. These molecular mechanisms illustrate how Pb can disrupt NO function in the brain, leading to various neurotoxic effects. Reduced NO availability and increased oxidative stress can impair synaptic plasticity, which is critical for learning and memory [83]. Pb-induced oxidative damage and inflammation may exacerbate cognitive deficits and memory impairment, while disruption of redox balance and NO signalling increases susceptibility to oxidative stress and inflammatory responses. Understanding these pathways highlights the complex interactions between Pb exposure and NO dysfunction and underscores the need for targeted strategies to mitigate the neurotoxic effects of lead [84].
ACh is known to have a dual function as both a neurotransmitter and a modulator of blood flow through endothelial cells [33]. This is important because NO produced by the endothelium in response to acetylcholine not only dilates blood vessels but also has neuroprotective effects by reducing oxidative stress and preventing inflammatory processes [13]. This mechanism is important not only in the local regulation of cerebral blood flow but also potentially in the broader context of vascular responses throughout the body [27]. In addition, in vivo evidence suggests that acetylcholine may influence pain modulation and sympathetic nervous system activity through NO-related mechanisms. For example, ACh released at the spinal level can induce antinociceptive effects and increase sympathetic activity, which may be related to NO synthesis and action. This is important for understanding how different parts of the nervous system work together to regulate both blood flow and pain responses, which may have clinical implications for the treatment of pain and circulatory disorders [31]. In the context of lead exposure, this relationship between acetylcholine and nitric oxide may be particularly significant. Lead exposure is known to disrupt both endothelial function and neurotransmitter signalling, potentially impairing NO production and the associated vasodilatory response [30, 85]. This disruption may lead to reduced cerebral blood flow, contributing to neurological deficits commonly seen in lead poisoning, such as cognitive impairment and neurodevelopmental delay [9]. In our study, we analysed the ACh-NO pathway, which may provide insights into mitigation of these effects by targeting endothelial dysfunction and increasing NO production [33], thereby improving blood flow and reducing the neurotoxic effects of lead nitrate exposure.
Zhong et al. [80] suggested that the involvement of both N-acetyl-L-cysteine and L-arginine in attenuating ROS generation highlights the importance of NOS activity in counteracting oxidative damage and showed that Pb exposure comprehensively disrupts NOS function. Indeed, this study highlighted that PbCl₂ treatment results in reduced expression of three different NOS isoforms – neuronal nitric oxide synthase (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS) at both transcriptional and translational levels in MCF-7 cells. A possible mechanism was the phosphorylation of eukaryotic initiation factor 2 (eIF2α), indicating stress responses leading to impaired cell function and survival. The association between Pb-induced oxidative stress and eIF2α phosphorylation highlights a potential mechanism by which Pb contributes to cellular dysfunction and cytotoxicity [80].
Kumawat et al. [86] also showed that Pb exposure affects microglia, the immune cells of the brain, by inducing them to secrete cytochemokines, which subsequently leads to neuronal death. Specifically, stimulation of BV-2 mouse microglia activates key signalling pathways, including extracellular signal-regulated kinase and protein kinase B, triggers the nuclear factor-κB transcription factor, and leads to increased levels of pro-inflammatory cytokines and enzymes, such as tumour necrosis factor-α, interleukin-6, monocyte chemoattractant protein-1, and cyclooxygenase-2, which contribute to the inflammatory response and neuronal damage associated with Pb exposure [86].
Our research has practical implications and shows how the findings can be applied to real-world scenarios. Since we administered lead orally to the rats in our study, the role of L-arginine becomes highly relevant in the context of lead intoxication. L-arginine is known to be a precursor for the production of NO, which plays a critical role in several physiological processes, including those involved in the body’s response to toxic substances, such as lead [87]. The presence of L-arginine in the diet can influence the synthesis of NO, which may modulate the severity of lead-induced oxidative stress and its effects on neuronal tissue [42]. In particular, the study highlights the potential benefits of L-arginine in mitigating the adverse effects of lead nitrate exposure. By demonstrating the influence of L-arginine on oxidative stress and subsequent neuronal damage associated with lead nitrate exposure, our findings provide valuable insights that could inform dietary recommendations and therapeutic strategies aimed at reducing the effects of lead nitrate poisoning [44]. This practical aspect highlights the relevance of our findings for public health interventions and the development of effective measures to protect individuals at risk of lead exposure.
Secondly, we investigated the oxidative modification of proteins and its role in lead-induced neurotoxicity, as lead exposure significantly affects neuronal protein structure and function and contributes to neuronal damage through increased aggregation, loss of function, and activation of apoptotic pathways [9]. Oxidative modification of proteins is a critical process that affects cellular function, particularly in neurons, where lead-induced oxidative stress leads to excessive production of ROS and reactive nitrogen species (RNS). These reactive molecules, including hydrogen peroxide (H₂O₂), hydroxyl radicals (∙OH), and singlet oxygen (¹O₂), can damage proteins, lipids, and DNA. In neuronal cells, which are particularly sensitive to oxidative stress, ROS can modify amino acids in proteins, e.g. oxidising tyrosine, tryptophan, and cysteine residues, resulting in an altered protein structure and function [19]. Examples of these modifications include tyrosine nitration and tryptophan nitrosylation.
The importance of L-arginine is particularly pronounced in individuals exposed to lead. As lead intoxication often leads to increased oxidative stress and disruption of cellular homeostasis, adequate levels of L-arginine may help to mitigate some of the harmful effects by supporting NO production. This in turn could help counteract the oxidative damage caused by lead exposure, potentially providing a protective mechanism against its neurotoxic effects. Thus, understanding the interaction between dietary L-arginine and lead toxicity is essential for developing strategies to manage and reduce the effects of lead exposure [44]. L-arginine also plays an important role in protecting against lead-induced oxidative stress by activating the enzymatic antioxidant defence. In our studies, we observed a statistically significant correlation between TBARS levels and GR activity (r = 0.768, p < 0.05) and OMP ADa and TAS (r = 0.813, p < 0.05) in the LR animals as well as DC and TAS (r = 0.611, p < 0.05) and CAT and TBARS (r = 0.821, p < 0.05) in the HR rats when L-arginine was administered prior to the lead nitrate exposure.
The study of diene conjugate (DC) and TBARS levels is key to understanding the effect of lead nitrate on brain tissue depending on the individual physiological characteristics of the organism, as brain tissue is characterised by its high lipid content. Lead can cause significant damage to neural tissue through the induction of oxidative stress and lipid peroxidation, with serious consequences for the function of the nervous system. Therefore, monitoring these indicators allows assessing the degree of neurotoxicity and understanding the mechanisms by which lead nitrate affects brain health; this is the approach used in the initial stages of our study. Based on our results, the effects of L-arginine and L-NNA on the levels of oxidatively modified proteins (OMPs) are markedly different and depend on the initial resistance to hypoxia (Figures 4 and 5). L-arginine, a nitric oxide precursor, did not significantly alter the OMP ADn levels but showed statistically significant effects between the high and low hypoxia-resistant groups, whether administered before or after the lead nitrate exposure. In contrast, L-NNA, an inhibitor of nitric oxide synthase, led to a more pronounced reduction in the OMP levels in high hypoxia-resistant rats, with significant effects observed for OMP KDn, OMP KDa, and OMP ADa. These results suggest that L-arginine and L-NNA have different effects on oxidative protein modifications, with L-arginine being more beneficial in low hypoxia-resistant rats by reducing OMP levels, whereas the effects of L-NNA are more pronounced in high hypoxia-resistant rats, highlighting the importance of hypoxia resistance in mediating these effects. These dependencies were confirmed in our correlation analyses, where we observed a statistically significant correlation between TBARS and OMP KDn (r = 0.778, p < 0.05) as well as TAS and OMP ADa (r = 0.893, p < 0.05) in the HR animals, where L-NNA was administered prior to the lead nitrate exposure.
Furthermore, it is important to consider the results of a study conducted by Nowak et al. [88], which highlights the critical effects of Pb on specific brain regions that are integral to emotional regulation, memory, and learning, in particular the prefrontal cortex, hippocampus, and cerebellum. Importantly, these areas are essential for higher cognitive functions and emotional responses, highlighting the serious consequences of Pb-induced neurotoxicity on these critical brain functions. Some of the results of the study confirm the importance of the NO pathway and glutamatergic neurotransmission in brain development, neurotoxicity, and neurodegeneration. Pb interference with both NO signalling and glutamatergic neurotransmission further exacerbates neurotoxic effects, as NO is critical for several brain functions, including neurovascular regulation and synaptic plasticity, while glutamatergic neurotransmission is fundamental for cognitive processes, such as learning and memory. The authors’ findings highlight Pb effects that disrupt NO signalling and glutamatergic systems in order to develop effective strategies to mitigate Pb-induced neurotoxicity [89]. By understanding the mechanisms by which Pb affects these critical pathways, we can better address the cognitive and emotional deficits associated with lead exposure and design targeted interventions to protect and restore brain function, especially in children [7, 15].
Thirdly, this study is also innovative in that it combines several different approaches: it examines the effect of lead nitrate exposure on both early and late stages of lipid peroxidation, looks in detail at oxidative modifications of proteins using four OMP parameters (neutral aliphatic aldehyde and ketone dinitrophenyl hydrazones, alkaline aliphatic and ketone aldehyde dinitrophenyl hydrazones), and assesses the role of antioxidant enzyme activity and biomarkers of cellular energy metabolism in brain tissue as a function of individual physiological reactivity (Fig. 11). By integrating these factors, the study aims to provide insight into lead-induced neurotoxicity and a deeper understanding of the interplay between oxidative stress and individual characteristics of organisms in the functioning of the nitric oxide system. In addition, oxidative damage can lead to loss of protein function, particularly affecting enzymatic and receptor proteins that are essential for neuronal activity. For example, modifications to chaperone proteins can impair their ability to properly fold and repair other proteins, exacerbating cellular stress. Oxidative modifications can also activate apoptotic pathways, accelerating neuronal cell death by affecting proteins involved in cell cycle regulation and apoptosis. This activation increases neurotoxicity and contributes to neuronal loss [20–22].
Fig. 11: Graphical abstract.
The results suggest that targeting nitric oxide pathways of metabolism may have different effects depending on the subject’s baseline resistance to hypoxia, particularly in the context of lead exposure. In animals, or potentially humans, with low resistance to hypoxia, L-arginine, a precursor for nitric oxide synthesis, may offer protective benefits against oxidative damage induced by lead nitrate exposure. This is evidenced by its ability to reduce the levels of oxidatively modified proteins in rats with low hypoxia resistance. Conversely, in those with high hypoxia resistance, the inhibition of nitric oxide synthesis with L-NNA may be more effective in reducing oxidative modifications. This is supported by the marked reduction in the OMP levels observed in the HR rats following the L-NNA administration, both before and after the lead nitrate exposure (Figures 4 and 5). Thus, therapeutic strategies involving modulation of the nitric oxide system should be tailored to individual’s resistance to hypoxia in order to optimise protection against lead-induced oxidative stress.
A study conducted by Reddy et al. [7] showed that low-level perinatal exposure to lead induces significant changes in the cholinergic system in the cerebellum and hippocampus of the developing brain. These changes persist even after Pb exposure is removed and may contribute to long-term behavioural and learning deficits. The cholinergic system, which is critical for neurotransmission and cognitive processes, is profoundly affected by Pb, leading to disruptions in cholinergic signalling pathways [90]. These disruptions can impair synaptic plasticity, memory formation, and learning abilities, highlighting the persistent effects of Pb on brain function and the importance of addressing Pb-induced neurotoxicity to mitigate its lasting effects on cognitive development [91]. Zhang et al. [92] showed in the developmental Japanese quail (Coturnix japonica ) model that cerebellar neurotransmission was disrupted by Pb, with increased ACh and decreased acetylcholinesterase activity, dopamine levels, and γ-aminobutyric acid (GABA) and Na+/K+-ATPase activity.
Many researchers have investigated the effects of lead-induced neurotoxicity on the cholinergic system of the brain, and their results differed due to differences in experimental models, doses, and conditions. Lead exposure may selectively affect the cholinergic system in different brain areas [36]. In studies using Drosophila , lead acetate exposure (0-20 mM for 7 days) significantly increased AChE activity and dopamine levels, as reported by Venkareddy and Muralidhara [35]. These studies also assessed lead-induced lethality, hyperactivity, oxidative stress, and neurotoxicity. In contrast, an in vitro study using the brains of 35-day-old rats showed a significant decrease in AChE activity at high Pb concentrations (50-100 µM), indicating a concentration-dependent effect [7]. Subsequent research carried out by Reddy et al. [36] showed that Pb exposure resulted in a significant decrease in AChE activity and an increase in ACh levels in the synaptosomal fractions of the cerebellum, hippocampus, and cerebral cortex. These effects were observed to be dose- and age-dependent, with the most pronounced changes occurring in the brains of young rats, particularly in the hippocampus. Furthermore, Basha et al. [34] investigated the effects of perinatal Pb exposure (0.2% lead acetate in maternal drinking water) on synaptosomal levels of ACh, epinephrine, dopamine, AChE, and mitochondrial monoamine oxidase (MAO) in the cerebellum and hippocampus of rats. This study found significant decreases in AChE and MAO activities and increased levels of ACh, dopamine, and epinephrine. In particular, these changes were more pronounced in the cholinergic system of the hippocampus and the aminergic system of the cerebellum. Thus, these studies demonstrate that lead exposure has complex and variable effects on neurotransmitter systems influenced by such factors as the animal model type, lead dose, and experimental conditions. This variability highlights the importance of careful consideration of these factors when interpreting the effects of lead-induced neurotoxicity. In addition, the effects of Pb exposure may vary significantly depending on the specific experimental conditions and the age of the subjects. Studies in rats have shown that Pb exposure reduces AChE activity and increases ACh levels in several brain regions, with more pronounced effects observed in younger animals. In contrast, other studies have reported different results depending on the age and brain region examined [36]. This variation highlights the importance of taking these factors into account when interpreting the effects of Pb toxicity.
The results of a study conducted by Liu et al. [93] show that puerarin, a natural flavonoid, inhibits lead-induced neurotoxicity by suppressing oxidative stress, reversing lead-induced changes in neurotransmitters and enzymes, and modulating the PKA/Akt/NOS signalling pathway in mice. Puerarin administration reduced Pb levels in both blood and brain via improved brain function by increasing AChE and MAO activities and by counteracting oxidative stress, as shown by reduced lipid peroxidation and restored total antioxidant capacity [19]. In addition, puerarin enhanced nitric oxide production and protein kinase A (PKA) activity, and increased the expression of key NOS isoforms and phosphorylated Akt. Another study conducted by Phyu and Tangpong [94] found that Pb induced AChE dysfunction and memory deficits in a dose-dependent manner, as demonstrated by both in vitro and in vivo experiments.
In this context, the results of the study conducted by Liu et al. [95] showed that lead induced apoptosis in kidney cells by triggering pro-apoptotic signalling pathways, as Pb exposure leads to the activation of apoptotic processes, including the release of cytochrome c from mitochondria and an imbalance between pro- and anti-apoptotic Bcl-2 proteins, promoting cell death. Puerarin, like other natural and synthetic antioxidants [93, 96, 97], counteracts these effects by increasing phosphorylated Akt and phosphorylated endothelial nitric oxide synthase (eNOS), which in turn increases renal nitric oxide levels, and this modulation of the PI3K/Akt/eNOS pathway helps to inhibit Pb-induced apoptosis.
Fourthly, overall, this research fills a gap in the literature and has the potential to advance our knowledge of neurotoxic mechanisms triggered by lead nitrate with dependence on the ACh-AChE system and the NO system. The findings may lead to novel therapeutic strategies and interventions, thereby improving our ability to manage and reduce the adverse effects of lead-induced toxicity. Our study has shown that the higher adaptive properties observed in the functional systems of HR animals compared to LR animals are crucial for understanding the fundamental differences in their responses to different stressors. The lead-induced exposure resulted in a statistically significant reduction in the TAS levels, with more pronounced effects in the highly resistant rats. The L-arginine administration resulted in a significant increase in the TAS levels in the LR and HR rats both before and after the lead nitrate exposure, with notable differences between the LR and HR animals (Fig. 8). In the brain tissue of the LR animals, L-NNA and the lead nitrate exposure abolished the beneficial effects of L-arginine. Conversely, the TAS levels in the HR animals were higher when L-NNA was administered after the lead nitrate exposure, compared to data obtained in the lead-exposed group alone. In this context, we observed a statistically significant correlation between TBARS and TAS (r = 0.768, p < 0.05) as well as AChE and TAS (r = 0.872, p < 0. 05) in the LR animals when L-arginine was administered prior to the lead nitrate exposure, and there were similar dependencies between ACh and DC levels (r = 0.568, p < 0.05), AChE and GR activity (r = 0.711, p < 0.05), and ACh and TAS (r = 0.841, p < 0.05) in the HR rats when L-NNA was administered after the lead nitrate exposure.
In addition to the above, a study conducted by Liu et al. [97] found that the protective effect of quercetin against Pb-induced neurotoxicity in mice exposed to lead acetate (20 mg/kg b.w. per day for 3 months) was mediated by a significant reduction in blood and brain lead levels (by 13.2% to 20.0%). It is important to note that antioxidants help restore critical lead-disrupted signalling pathways involved in cell survival, neurotransmission, and cognitive function [81]. Natural antioxidant products, such as quercetin, increased the phosphorylation of several key proteins, including Akt, Ca²⁺/calmodulin-dependent protein kinase II (CaMKII), neuronal nitric oxide synthase (nNOS), endothelial nitric oxide synthase (eNOS), and cAMP response element-binding protein (CREB) [97]. Highlighting the protective effects of selenium against oxidative stress and neuronal damage, research by Hegazy et al. [98] investigated the potential role of selenium in mitigating lead-induced neurotoxicity in the cerebrum of adult male rats. This experimental study provides valuable insights into the mechanisms by which selenium may promote brain health and has promising implications for therapeutic strategies in cases of lead exposure.
Given the variability in the response of different individuals to stress and disease based on their physiological reactivity, personalised approaches may offer more precise and effective therapeutic options. In addition, the effect of heavy metals in exacerbating stress and the dependence of this effect on individual physiological reactivity highlights the need for a comprehensive approach. Heavy metals can interfere with NO production and other biochemical and physiological processes, further affecting stress responses and disease progression [12, 80, 84]. Our previous research [40, 44] suggests that L-arginine-based interventions may improve the adaptive characteristics of LR animals. Male animals were used instead of females to avoid potential influences of estrous cycle hormones on the study results [99, 100].
Fifthly, we observed significant increases in alanine and aspartate aminotransferase activity in the brain tissue after the lead nitrate exposure, with more pronounced effects when L-arginine was administered before the lead nitrate exposure. The administration of the nitric oxide synthase inhibitor L-NNA reversed these changes. The de Ritis coefficient, which is used to assess the prognosis of pathological changes, showed that L-arginine reduced this coefficient, compared to the data obtained with the lead nitrate exposure alone, and further reductions were observed with the L-NNA treatment (Table 1). In addition, the lead nitrate exposure affected the activities of SDH and KGDH, which are critical for cellular energy production and resistance to hypoxia. The administration of L-arginine prior to the lead nitrate exposure altered the SDH activity differently in high and low resistance animals, whereas L-NNA increased SDH and KGDH activities in both groups, suggesting potential therapeutic implications of nitric oxide system modulators in mitigating lead-induced damage. In our studies, we observed a statistically significant correlation between TBARS and SDH (r = 0.758, p < 0.05), TAS and KGDH (r = 0.698, p < 0.05), and OMP ADa and SDH (r = 0.828, p < 0.05) in the LR group during the lead nitrate exposure. In the group of HR animals, significant correlations were shown between the ALT and SDH activity (r = 0.713, p < 0.05) and AST and TAS (r = 0.518, p < 0.05) in similar conditions of lead nitrate exposure.
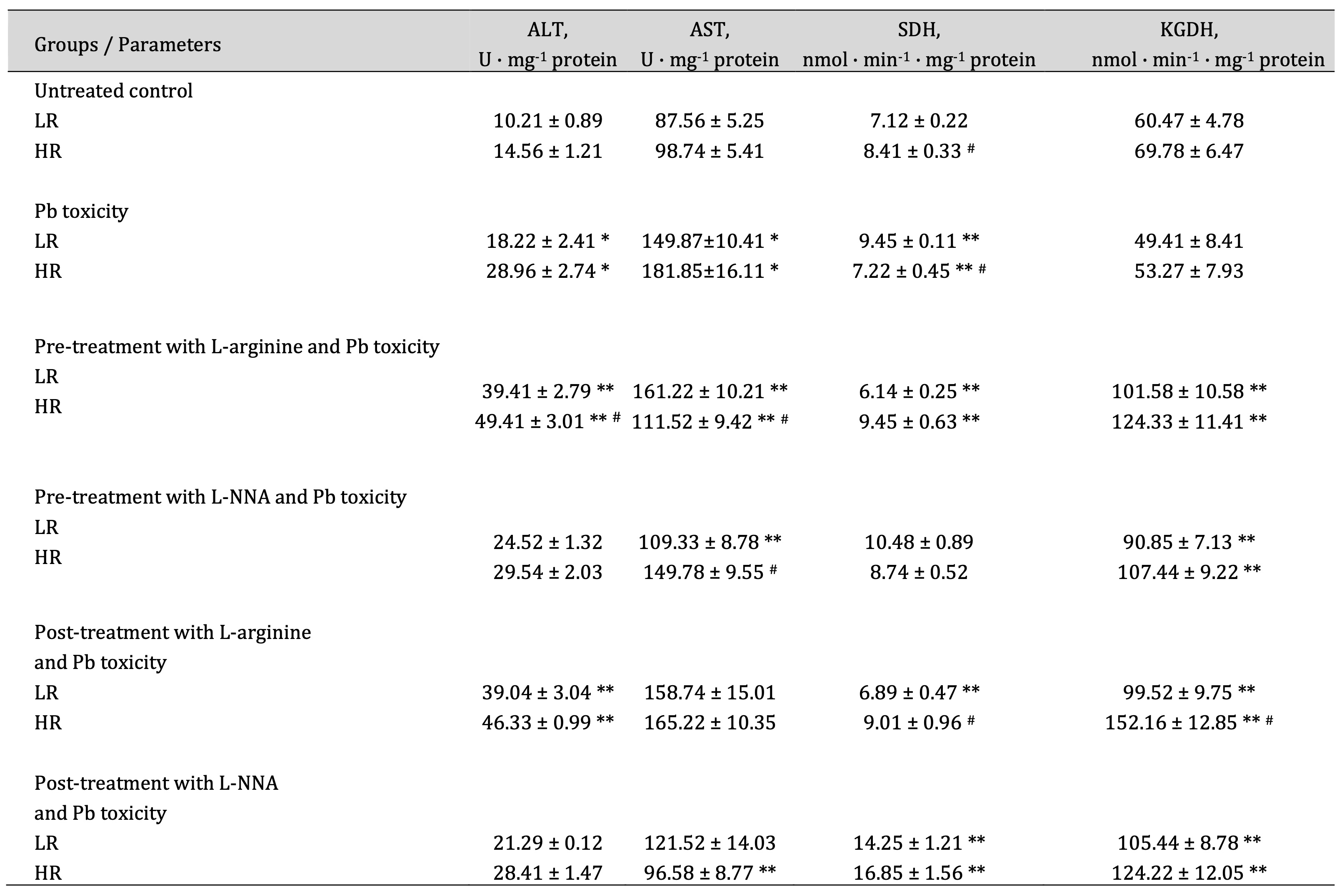
Table 1: Activities of alanine aminotransferase (ALT, U · mg-1 protein), aspartate aminotransferase (AST, U · mg-1 protein), succinate dehydrogenase (SDH, nmol · min-1 · mg-1 protein), and α-ketoglutarate dehydrogenase (KGDH, nmol · min-1· mg-1 protein) in the brain tissue of rats with low (LR) and high (HR) hypoxia resistance exposed to lead nitrate for 30 days and treated with L-arginine (600 mg/kg, 30 min) or L-NNA (35 mg/kg, 30 min) before and after the exposure to lead nitrate (n = 6). Data are expressed as mean ± S.D. (n = 6). * Significant differences between the Pb group and the untreated control (p < 0.05). ** Significant differences between the Pb group and the L-arginine + Pb group, the L-NNA + Pb group, the Pb + L-arginine group, and the Pb + L-NNA group (p < 0.05). # Significant differences between the LR and HR groups in the same conditions (p < 0.05)

We found that lead poisoning significantly affects glutamate metabolism and key Krebs cycle enzymes. Lead disrupts the function of ALT and AST, leading to reduced levels of glutamate, which is a key precursor of α-ketoglutarate. Alpha-ketoglutarate is a key compound in the Krebs cycle, where it is converted to succinyl-CoA by α-ketoglutarate dehydrogenase. Disruption of this process results in reduced ATP production, which is detrimental to neuronal function. These disturbances depend on the individual’s resistance to hypoxia, as different levels of resistance can affect the extent of metabolic disturbances. Lead nitrate exposure directly inhibits the activity of SDH and acts on KGDH, leading to the accumulation of succinate and a further reduction in energy production (Table 1). SDH inhibition affects both the Krebs cycle and the electron transport chain, exacerbating ATP deficits and damaging cells [101]. In addition, exposure to lead nitrate increases the production of ROS, further damaging these enzymes and exacerbating energy dysfunction. The impact of these processes varies with the level of initial hypoxia resistance and influences the manifestation and progression of lead-induced metabolic disorders.
The main strength of this study is the detailed examination of oxidative stress markers and antioxidant defences specifically in brain tissue exposed to lead nitrate. By focusing on biochemical indicators, such as lipid peroxidation and oxidative modification of proteins, and by assessing the activity of antioxidant enzymes, the study provides valuable insights into the biochemical changes occurring in brain tissue. The emphasis on individual physiological characteristics related to hypoxia resistance further enhances the relevance of the study, providing a more nuanced understanding of how different individuals might respond to lead-induced oxidative stress. This approach not only advances the knowledge of oxidative stress research, but also supports the development of targeted strategies to mitigate lead-induced oxidative damage based on individual biochemical and physiological profiles.
Despite its strengths, the study has several limitations. The primary limitation is the exclusive focus on brain tissue, which may not fully capture the broader systemic effects of oxidative stress and biochemical responses caused by lead nitrate exposure in different organs. In addition, the research did not address the immunological aspects of oxidative stress, such as the effects of lead exposure on inflammatory responses and immune cell function. Future research will address these gaps by including a wider range of tissues and integrating immunological assessments. In addition, the inclusion of genetic analyses will be critical to understanding the influence of genetic variability on biochemical and immunological responses to lead-induced oxidative stress, thereby improving the precision and applicability of the findings.
This study provides direction for future research by highlighting the need to further investigate the biochemical and immunological aspects of oxidative stress in the brain caused by lead nitrate exposure. Future studies will focus on the detailed biochemical pathways involved in lead-induced lipid peroxidation and protein damage, and the role of antioxidant enzymes in different tissues of individuals with different resistance to hypoxia. It will be important to investigate how these biochemical processes depend on the initial physiological reactivity of organisms and how these processes are modulated by the NO system. In addition, incorporating assessments of baseline physiological reactivity to evaluate the effect of lead nitrate exposure on metabolic responses in different tissues may provide a more complete understanding of the systemic effects of lead-induced oxidative stress and therapeutic approaches using modulators of the NO system.
Conclusion
The results of this study suggest that the effectiveness of modifying nitric oxide pathways in counteracting oxidative damage in brain tissue of rats exposed to lead nitrate, assessed by measuring biomarkers of lipoperoxidation, protein damage, antioxidant enzyme activity, the acetylcholine-cholinesterase system, varies with individual hypoxia resistance. In rats with low resistance to hypoxia, L-arginine, which increases nitric oxide production, may provide a protective benefit by reducing lead-induced oxidative stress. This is evidenced by its ability to reduce the levels of carbonyl derivatives of oxidatively modified proteins and to attenuate lipoperoxidation in animals with low hypoxia resistance. In addition, L-arginine may support the activity of antioxidant enzymes, helping to neutralise oxidative stress. Conversely, in rats with high hypoxia resistance, L-NNA, an inhibitor of nitric oxide synthesis, may be more effective in reducing oxidative modifications, as shown by the significant reduction in the levels of oxidatively modified proteins and lipoperoxidation. L-NNA also appears to affect the acetylcholine-cholinesterase system, potentially altering neurotransmitter balance and further influencing oxidative stress responses. Therefore, therapeutic strategies involving modulation of the nitric oxide system may control lipoperoxidation, support the activity of antioxidant enzymes, and regulate the acetylcholine-cholinesterase system. This approach should take into account individual’s resistance to hypoxia in order to effectively address oxidative stress caused by lead nitrate exposure.
Abbreviations
Central nervous system – CNS; lead – Pb; nitric oxide – NO; neuronal nitric oxide synthase – nNOS; endothelial nitric oxide synthase - eNOS; inducible nitric oxide synthase – iNOS; reactive oxygen species – ROS; reactive nitrogen species – RNS; acetylcholine – Ach; muscarinic M5 receptors – M5-mAchRs; acetylcholinesterase – AChE; mitochondrial monoamine oxidase – MAO; Nω-nitro-L-arginine - L-NNA; diene conjugates – DC; 2-thiobarbituric acid reactive substances – TBARS; malonic dialdehyde – MDA; oxidatively modified proteins – OMP; neutral aldehyde dinitrophenylhydrazones – OMP ADn; neutral aliphatic ketone dinitrophenylhydrazones – OMP KDn; alkaline aliphatic aldehyde dinitrophenylhydrazones – OMP ADa; alkaline aliphatic ketone dinitrophenylhydrazones –OMP KDa; superoxide dismutase – SOD; catalase – CAT; glutathione reductase – GR; glutathione peroxidase – GPx; total antioxidant status – TAS; alanine aminotransferase – ALT; aspartate aminotransferase – AST; succinate dehydrogenase – SDH; α-ketoglutarate dehydrogenase – KGDH; low – (LR) and high – (HR) resistance to hypoxia.
Acknowledgements
Authors contributions
The authors contributed to the following aspects of the study Conceptualisation: HT, NK, OL, PK; Data curation: HT, NK; Formal analysis: HT, NK; Investigation: HT, NK, OL; Methodology: HT, NK; Supervision: NK, HT; Writing – original draft: NK, HT; Writing – revision and editing: HT, NK, PK.
Natalia Kurhaluk, ORCID: https://orcid.org/0000-0002-4669-1092;
Oleksandr Lukash, ORCID: https://orcid.org/0000-0003-2702-6430;
Piotr Kamiński, ORCID: https://orcid.org/0000-0003-1978-6018;
Halina Tkaczenko, ORCID: https://orcid.org/0000-0003-3951-9005
Funding
The present study was financially supported by the Pomeranian University in Słupsk (Słupsk, Poland) and T.H. Shevchenko National University “Chernihiv Colehium” (Chernihiv, Ukraine).
The present study was financially supported by the Pomeranian University in Słupsk (Słupsk, Poland) and T.H. Shevchenko National University “Chernihiv Colehium” (Chernihiv, Ukraine). The authors would like to thank the Pomeranian University in Słupsk and T.H. Shevchenko National University “Chernihiv Colehium” for supporting this research.
Disclosure Statement
The authors have no competing interests to declare.
References
| 1 | Andrade VM, Aschner M, Marreilha Dos Santos AP: Neurotoxicity of Metal Mixtures. Adv Neurobiol 2017;18:227-265.
https://doi.org/10.1007/978-3-319-60189-2_12 |
| 2 | Li B, Xia M, Zorec R, Parpura V, Verkhratsky A: Astrocytes in heavy metal neurotoxicity and neurodegeneration. Brain Res 2021;1752:147234.
https://doi.org/10.1016/j.brainres.2020.147234 |
| 3 | Mason LH, Harp JP, Han DY: Pb neurotoxicity: neuropsychological effects of lead toxicity. Biomed Res Int 2014;2014:840547.
https://doi.org/10.1155/2014/840547 |
| 4 | Bhasin T, Lamture Y, Kumar M, Dhamecha R: Unveiling the Health Ramifications of Lead Poisoning: A Narrative Review. Cureus. 2023; 15(10):e46727.
https://doi.org/10.7759/cureus.46727 |
| 5 | Wu X, Cobbina SJ, Mao G, Xu H, Zhang Z, Yang L: A review of toxicity and mechanisms of individual and mixtures of heavy metals in the environment. Environ Sci Pollut Res Int 2016;23(9):8244-8259.
https://doi.org/10.1007/s11356-016-6333-x |
| 6 | Karri V, Schuhmacher M, Kumar V: Heavy metals (Pb, Cd, As and MeHg) as risk factors for cognitive dysfunction: A general review of metal mixture mechanism in brain. Environ. Toxicol Pharmacol 2016;48:203-213.
https://doi.org/10.1016/j.etap.2016.09.016 |
| 7 | Reddy GR, Basha MR, Devi CB, Suresh A, Baker JL, Shafeek A, Heinz J, Chetty CS: Lead induced effects on acetylcholinesterase activity in cerebellum and hippocampus of developing rat. Int J Dev Neurosci 2003;21(6):347-352.
https://doi.org/10.1016/S0736-5748(03)00071-6 |
| 8 | Lee JW, Choi H, Hwang UK, Kang JC, Kang YJ, Kim KI, Kim JH. Toxic effects of lead exposure on bioaccumulation, oxidative stress, neurotoxicity, and immune responses in fish: A review. Environ Toxicol Pharmacol 2019;68:101-108.
https://doi.org/10.1016/j.etap.2019.03.010 |
| 9 | Sanders T, Liu Y, Buchner V, Tchounwou, PB: Neurotoxic effects and biomarkers of lead exposure: a review. Reviews on environmental health 2009;24(1):15-45.
https://doi.org/10.1515/REVEH.2009.24.1.15 |
| 10 | Ahmad F, Liu P: (Ascorb)ing Pb Neurotoxicity in the Developing Brain. Antioxidants (Basel) 2020;9(12):1311.
https://doi.org/10.3390/antiox9121311 |
| 11 | Costa LG, Aschner M, Vitalone A, Syversen T, Soldin OP: Developmental neuropathology of environmental agents. Annu Rev Pharmacol Toxicol 2004;44:87-110.
https://doi.org/10.1146/annurev.pharmtox.44.101802.121424 |
| 12 | Nava-Ruiz C, Méndez-Armenta M, Ríos C: Lead neurotoxicity: effects on brain nitric oxide synthase. J Mol Histol 2012;43(5):553-563.
https://doi.org/10.1007/s10735-012-9414-2 |
| 13 | McCann SM, Kimura M, Karanth S, Yu WH, Rettori V: Nitric oxide controls the hypothalamic-pituitary response to cytokines. Neuroimmunomodulation 1997;4(2):98-106.
https://doi.org/10.1159/000097327 |
| 14 | Vaziri ND, Ding Y, Ni Z: Nitric oxide synthase expression in the course of lead-induced hypertension. Hypertension 1999;34(4 Pt. 1):558-562.
https://doi.org/10.1161/01.HYP.34.4.558 |
| 15 | Chetty CS, Reddy GR, Murthy KS, Johnson J, Sajwan K, Desaiah D. Perinatal lead exposure alters the expression of neuronal nitric oxide synthase in rat brain. Int J Toxicol 2001;20(3):113-120.
https://doi.org/10.1080/109158101317097692 |
| 16 | Chibowska K, Baranowska-Bosiacka I, Falkowska A, Gutowska I, Goschorska M, Chlubek D: Effect of Lead (Pb) on Inflammatory Processes in the Brain. Int J Mol Sci 2016;17(12):2140.
https://doi.org/10.3390/ijms17122140 |
| 17 | Karri V, Kumar V, Ramos D, Oliveira E, Schuhmacher M: Comparative In vitro Toxicity Evaluation of Heavy Metals (Lead, Cadmium, Arsenic, and Methylmercury) on HT-22 Hippocampal Cell Line. Biol Trace Elem Res 2018;184(1):226-239.
https://doi.org/10.1007/s12011-017-1177-x |
| 18 | Gundacker C, Forsthuber M, Szigeti T, Kakucs R, Mustieles V, Fernandez MF, Bengtsen E, Vogel U, Hougaard KS, Saber AT: Lead (Pb) and neurodevelopment: A review on exposure and biomarkers of effect (BDNF, HDL) and susceptibility. Int J Hyg Environ Health 2021;238:113855.
https://doi.org/10.1016/j.ijheh.2021.113855 |
| 19 | Soratto Heitich Ferrazza MH, Delwing-Dal Magro D, Salamaia E, Guareschi TE, Fernandes Erzinger LF, Maia TP, Siebert C, Dos Santos TM, de Souza Wyse AT, Borgmann G, Plautz K, Delwing-de Lima D: Sub chronic administration of lead alters markers of oxidative stress, acetylcholinesterase and Na+K+ ATPase activities in rat brain. Acta Neurobiol Exp (Wars) 2023;83(2):216-225.
https://doi.org/10.55782/ane-2023-019 |
| 20 | Cobb CA, Cole MP: Oxidative and nitrative stress in neurodegeneration. Neurobiol Dis 2015;84:4-21.
https://doi.org/10.1016/j.nbd.2015.04.020 |
| 21 | Morris G, Walder K, Puri BK, Berk M, Maes M. The Deleterious Effects of Oxidative and Nitrosative Stress on Palmitoylation, Membrane Lipid Rafts and Lipid-Based Cellular Signalling: New Drug Targets in Neuroimmune Disorders. Mol Neurobiol 2016;53(7):4638-4658.
https://doi.org/10.1007/s12035-015-9392-y |
| 22 | Valko M, Jomova K, Rhodes CJ, Kuča K, Musílek K: Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch Toxicol 2016;90(1):1-37.
https://doi.org/10.1007/s00204-015-1579-5 |
| 23 | Pounds JG: Effect of lead intoxication on calcium homeostasis and calcium-mediated cell function: a review. Neurotoxicology 1984;5(3):295-331.
|
| 24 | Esplugues JV: NO as a signalling molecule in the nervous system. Br J Pharmacol 2002;135(5):1079-1095.
https://doi.org/10.1038/sj.bjp.0704569 |
| 25 | Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB: Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 2014;20(7):1126-1167.
https://doi.org/10.1089/ars.2012.5149 |
| 26 | Franzoni F, Scarfò G, Guidotti S, Fusi J, Asomov M, Pruneti C: Oxidative Stress and Cognitive Decline: The Neuroprotective Role of Natural Antioxidants. Front Neurosci 2021;15:729757.
https://doi.org/10.3389/fnins.2021.729757 |
| 27 | Xu Z, Tong C, Eisenach JC: Acetylcholine stimulates the release of nitric oxide from rat spinal cord. Anesthesiology 1996;85(1):107-111.
https://doi.org/10.1097/00000542-199607000-00015 |
| 28 | Zuccolo E, Laforenza U, Negri S, Botta L, Berra-Romani R, Faris P, Scarpellino G, Forcaia G, Pellavio G, Sancini G, Moccia F. Muscarinic M5 receptors trigger acetylcholine-induced Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. J Cell Physiol 2019;234(4):4540-4562.
https://doi.org/10.1002/jcp.27234 |
| 29 | Ramírez Ortega D, González Esquivel DF, Blanco Ayala T, Pineda B, Gómez Manzo S, Marcial Quino J, Carrillo Mora P, Pérez de la Cruz V: Cognitive Impairment Induced by Lead Exposure during Lifespan: Mechanisms of Lead Neurotoxicity. Toxics 2021;9(2):23.
https://doi.org/10.3390/toxics9020023 |
| 30 | Vaziri ND: Mechanisms of lead-induced hypertension and cardiovascular disease. American journal of physiology. Heart and circulatory physiology 2008;295(2):H454-H465.
https://doi.org/10.1152/ajpheart.00158.2008 |
| 31 | Minatel E, Santo Neto H, Marques MJ: Acetylcholine receptors and neuronal nitric oxide synthase distribution at the neuromuscular junction of regenerated muscle fibers. Muscle Nerve 2001;24(3):410-416.
https://doi.org/10.1002/1097-4598(200103)24:3<410::AID-MUS1014>3.3.CO;2-S |
| 32 | Elhusseiny A, Hamel E: Muscarinic - but not nicotinic - acetylcholine receptors mediate a nitric oxide-dependent dilation in brain cortical arterioles: a possible role for the M5 receptor subtype. J Cereb Blood Flow Metab 2000;20(2):298-305.
https://doi.org/10.1097/00004647-200002000-00011 |
| 33 | Zuccolo E, Lim D, Kheder DA, Perna A, Catarsi P, Botta L, Rosti V, Riboni L, Sancini G, Tanzi F, D'Angelo E, Guerra G, Moccia F. Acetylcholine induces intracellular Ca2+ oscillations and nitric oxide release in mouse brain endothelial cells. Cell Calcium 2017;66:33-47.
https://doi.org/10.1016/j.ceca.2017.06.003 |
| 34 | Basha DC, Rani MU, Devi CB, Kumar MR, Reddy GR: Perinatal lead exposure alters postnatal cholinergic and aminergic system in rat brain: reversal effect of calcium co-administration. Int J Dev Neurosci 2012;30(4):343-350.
https://doi.org/10.1016/j.ijdevneu.2012.01.004 |
| 35 | Venkareddy LK, Muralidhara: Potential of casein as a nutrient intervention to alleviate lead (Pb) acetate-mediated oxidative stress and neurotoxicity: First evidence in Drosophila melanogaster. Neurotoxicology 2015;48:142-151.
https://doi.org/10.1016/j.neuro.2015.03.014 |
| 36 | Reddy GR, Devi BC, Chetty CS: Developmental lead neurotoxicity: alterations in brain cholinergic system. Neurotoxicology 2007;28(2):402-407.
https://doi.org/10.1016/j.neuro.2006.03.018 |
| 37 | Puntel RL, Nogueira CW, Rocha JB: Krebs cycle intermediates modulate thiobarbituric acid reactive species (TBARS) production in rat brain in vitro. Neurochem Res 2005;30(2):225-235.
https://doi.org/10.1007/s11064-004-2445-7 |
| 38 | Ryan DG, Murphy MP, Frezza C, Prag HA, Chouchani ET, O'Neill LA, Mills EL: Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat Metab 2019;1:16-33.
https://doi.org/10.1038/s42255-018-0014-7 |
| 39 | Murphy MP, O'Neill LAJ: Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 2018;174(4):780-784.
https://doi.org/10.1016/j.cell.2018.07.030 |
| 40 | Kurhaluk N, Lukash O, Tkaczenko H: Do the Effects of Krebs Cycle Intermediates on Oxygen-Dependent Processes in Hypoxia Mediated by the Nitric Oxide System Have Reciprocal or Competitive Relationships? Cell Physiol Biochem 2023;57(6):426-451.
https://doi.org/10.33594/000000669 |
| 41 | Kurhaluk N, Lukash O, Kamiński P, Tkaczenko H: Adaptive Effects of Intermittent Hypoxia Training on Oxygen-Dependent Processes as a Potential Therapeutic Strategy Tool. Cell Physiol Biochem 2024;58(3):226-249.
https://doi.org/10.33594/000000705 |
| 42 | Kurhaluk N: The Effectiveness of L-arginine in Clinical Conditions Associated with Hypoxia. Int J Mol Sci 2023;24(9):8205.
https://doi.org/10.3390/ijms24098205 |
| 43 | Kurhaluk N: Supplementation with L-arginine and nitrates vs age and individual physiological reactivity. Nutr Rev 2024;82(9):1239-1259.
https://doi.org/10.1093/nutrit/nuad131 |
| 44 | Tkaczenko H, Lukash O, Kamiński P, Kurhaluk N: Elucidation of the Role of L-Arginine and Nω-Nitro-L-Arginine in the Treatment of Rats with Different Levels of Hypoxic Tolerance and Exposure to Lead Nitrate. Cell Physiol Biochem 2024;58:336-360.
https://doi.org/10.33594/000000716 |
| 45 | Lundberg JO, Weitzberg E: Nitric oxide signaling in health and disease. Cell 2022;185(16):2853-2878.
https://doi.org/10.1016/j.cell.2022.06.010 |
| 46 | Dzhalilova DS, Zolotova NA, Mkhitarov VA, Kosyreva AM, Tsvetkov IS, Khalansky AS, Alekseeva AI, Fatkhudinov TH, Makarova OV: Morphological and molecular-biological features of glioblastoma progression in tolerant and susceptible to hypoxia Wistar rats. Sci Rep 2023;13(1):12694.
https://doi.org/10.1038/s41598-023-39914-9 |
| 47 | Dzhalilova DS, Silina MV, Kosyreva AM, Tsvetkov IS, Makarova OV: Morphological and Molecular Biological Features of the Systemic Inflammatory Response in Old Wistar Rats with High and Low Resistance to Hypoxia. Bull Exp Biol Med 2023;175(5):704-710.
https://doi.org/10.1007/s10517-023-05930-y |
| 48 | Dzhalilova DS, Kosyreva AM, Tsvetkov IS, Zolotova NA, Sentyabreva AV, Makarova OV: Morphofunctional Changes in the Thymus in Prepubertal Male Wistar Rats in LPS-Induced Systemic Inflammatory Response in Relation to Hypoxia Tolerance. Bull Exp Biol Med 2023;174(3):385-390.
https://doi.org/10.1007/s10517-023-05713-5 |
| 49 | Kosyreva AM, Dzhalilova DS, Tsvetkov IS, Makarova MA, Makarova OV: Ex Vivo Production of IL-1β and IL-10 by Activated Blood Cells of Wistar Rats with Different Resistance to Hypoxia after Systemic Inflammatory Response Syndrome. Bull Exp Biol Med 2023;176(2):290-296.
https://doi.org/10.1007/s10517-024-06010-5 |
| 50 | Dzhalilova DS, Silina MV, Kosyreva AM, Tsvetkov IS, Makarova OV: Comparative Molecular and Biological Characteristic of the Systemic Inflammatory Response in Adult and Old Male Wistar Rats with Different Resistance to Hypoxia. Bull Exp Biol Med 2024;176(5):680-686.
https://doi.org/10.1007/s10517-024-06090-3 |
| 51 | Kurhaliuk NM, Serebrovs'ka TV, Koliesnikova IeE, Aleksiuk LI: Moduliatsiia ekzohennym L-arhininom mitokhondrial'noho ta mikrosomal'noho okysnennia pry hostriĭ ta periodychniĭ normobarychniĭ hipoksiï [Exogenous L-arginine modulates mitochondrial and microsomal oxidation in acute and intermittent normobaric hypoxia]. Fiziol Zh (1994) 2002;48(5):67-73.
|
| 52 | Kurhaliuk NM, Serebrovs'ka TV, Nosar VI, Kolesnikova EE, Moĭbenko OO: Interval'ni hipoksychni trenuvannia ta L-arhinin iak zasoby korektsiï enerhozabezpechennia miokarda za umov hostroï hipoksiï [Intermittent hypoxic training and L-arginine as corrective agents for myocardial energy supply under acute hypoxia]. Ukr Biokhim Zh (1999) 2002;74(1):82-87.
|
| 53 | Tkachenko H, Kurhalyuk N: Role of L-arginine Against Lead Toxicity in Liver of Rats with Different Resistance to Hypoxia. Pol J Environ Stud 2011;20(5):1319-1325.
|
| 54 | Bradford MM: A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248-254.
https://doi.org/10.1006/abio.1976.9999 |
| 55 | Kamyshnikov VS: Spectrophotometric determination of acyl hydroperoxides (diene conjugates) in blood plasma (serum). In: Reference book on clinical and biochemical research and laboratory diagnostics. Moscow, MEDpress-inform, 2009; pp. 548-549.
|
| 56 | Buege JA, Aust SD: Microsomal lipid peroxidation. Methods Enzymol 1978;52:302-310.
https://doi.org/10.1016/S0076-6879(78)52032-6 |
| 57 | Reznick AZ, Packer L: Oxidative damage to proteins: spectrophotometric method for carbonyl assay. Methods Enzymol 1994;233:357-363.
https://doi.org/10.1016/S0076-6879(94)33041-7 |
| 58 | Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz AG, Ahn BW, Shaltiel S, Stadtman ER: Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol 1990;186:464-478.
https://doi.org/10.1016/0076-6879(90)86141-H |
| 59 | Woolliams JA, Wiener G, Anderson PH, McMurray CH: Variation in the activities of glutathione peroxidase and superoxide dismutase and in the concentration of copper in the blood in various breed crosses of sheep. Res Vet Sci 1983;34(3):253-256.
https://doi.org/10.1016/S0034-5288(18)32219-7 |
| 60 | Suttle NF, McMurray CH: Use of erythrocyte copper:zinc superoxide dismutase activity and hair or fleece copper concentrations in the diagnosis of hypocuprosis in ruminants. Res Vet Sci 1983;35(1):47-52.
https://doi.org/10.1016/S0034-5288(18)32201-X |
| 61 | Koroliuk MA, Ivanova LI, Maĭorova IG, Tokarev VE: Metod opredeleniia aktivnosti katalazy [A method of determining catalase activity]. Lab Delo 1988;(1):16-19.
|
| 62 | Goldberg DM, Spooner RJ: Assay of Glutathione Reductase. In: Bergmeyen, H.V., Ed., Methods of Enzymatic Analysis, 3rd Edition, Vol. 3, Verlog Chemie, Deerfiled Beach, 1983, pp. 258-265.
|
| 63 | Melissinos KG, Delidou AZ, Varsou AG, Begietti SS, Drivas GJ: Serum and erythrocyte glutathione reductase activity in chronic renal failure. Nephron 1981;28(2):76-79.
https://doi.org/10.1159/000182115 |
| 64 | Paglia DE, Valentine WN: Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med 1967;70(1):158-169.
|
| 65 | Kraus RJ, Ganther HE: Reaction of cyanide with glutathione peroxidase. Biochem Biophys Res Commun 1980;96(3):1116-1122.
https://doi.org/10.1016/0006-291X(80)90067-4 |
| 66 | Miller NJ, Rice-Evans C, Davies MJ, Gopinathan V, Milner A: A novel method for measuring antioxidant capacity and its application to monitoring the antioxidant status in premature neonates. Clin Sci (Lond) 1993;84(4):407-412.
https://doi.org/10.1042/cs0840407 |
| 67 | Shutskiĭ IV: A method for determining acetylcholine in small amounts of blood. Lab Delo 1967;7:407-408.
|
| 68 | Menshikov VV: Laboratory methods of research in the clinic practice. Moscow, Medicine, 1987, pp. 225-227.
|
| 69 | Ellman GL, Courtney KD, Andres VJr, Feather-Stone RM: A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88-95.
https://doi.org/10.1016/0006-2952(61)90145-9 |
| 70 | Reitman S, Frankel S: A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. Am J Clin Pathol 1957;28(1):56-63.
https://doi.org/10.1093/ajcp/28.1.56 |
| 71 | Eschenko ND, Volski GG: Determination of succinic acid level and succinate dehydrogenase activity. In: Methods of biochemical study. Leningrad, Sankt-Petersburg National University, 1982; pp. 207-212.
|
| 72 | Eschenko ND, Volski GG: Determination of α-ketoglutaric acid level and α-ketoglutarate dehydrogenase activity. In: Methods of biochemical study. Leningrad, Sankt-Petersburg National University, 1982; pp. 202-206.
|
| 73 | Olufemi AC, Mji A, Mukhola MS: Potential Health Risks of Lead Exposure from Early Life through Later Life: Implications for Public Health Education. Int J Environ Res Public Health 2022;19(23):16006.
https://doi.org/10.3390/ijerph192316006 |
| 74 | Jomova K, Valko M: Advances in metal-induced oxidative stress and human disease. Toxicology 2011;283(2-3):65-87.
https://doi.org/10.1016/j.tox.2011.03.001 |
| 75 | Matović V, Buha A, Ðukić-Ćosić D, Bulat Z: Insight into the oxidative stress induced by lead and/or cadmium in blood, liver and kidneys. Food Chem Toxicol 2015;78:130-140.
https://doi.org/10.1016/j.fct.2015.02.011 |
| 76 | Lopes AC, Peixe TS, Mesas AE, Paoliello MM: Lead Exposure and Oxidative Stress: A Systematic Review. Rev Environ Contam Toxicol 2016;236:193-238.
https://doi.org/10.1007/978-3-319-20013-2_3 |
| 77 | Zimet Z, Bilban M, Fabjan T, Suhadolc K, Poljšak B, Osredkar J: Lead Exposure and Oxidative Stress in Coal Miners. Biomed Environ Sci 2017;30(11):841-845.
|
| 78 | Virgolini MB, Aschner M: Molecular mechanisms of lead neurotoxicity. Adv Neurotoxicol 2021;5:159-213.
https://doi.org/10.1016/bs.ant.2020.11.002 |
| 79 | Verstraeten SV, Aimo L, Oteiza PI: Aluminium and lead: molecular mechanisms of brain toxicity. Arch Toxicol 2008;82(11):789-802.
https://doi.org/10.1007/s00204-008-0345-3 |
| 80 | Zhong L, Wang L, Xu L, Liu Q, Jiang L, Zhi Y, Lu W, Zhou P: The cytotoxic effect of the NOS-mediated oxidative stress in MCF-7 cells after PbCl₂ exposure. Environ Toxicol 2016;31(5):601-608.
https://doi.org/10.1002/tox.22073 |
| 81 | Neal AP, Guilarte TR: Molecular neurobiology of lead (Pb(2+)): effects on synaptic function. Mol Neurobiol 2010;42(3):151-160.
https://doi.org/10.1007/s12035-010-8146-0 |
| 82 | Davtalab S, Karimi E, Moghaddam MN, Shokryazdan P, Jahromi MF, Oskoueian E: Biosorption and Bioprotective Potential of Levilactobacillus brevis in Mice Challenged by Lead-Induced Oxidative Stress. Biol Trace Elem Res 2024;202(11):5157-5165.
https://doi.org/10.1007/s12011-024-04080-0 |
| 83 | Reddy GR, Suresh A, Murthy KS, Chetty CS: Lead neurotoxicity: heme oxygenase and nitric oxide synthase activities in developing rat brain. Neurotox Res 2002;4(1):33-39 .
https://doi.org/10.1080/10298420290007600 |
| 84 | Nowak P, Swoboda M, Szkilnik R, Jośko J, Noras L, Kwieciński A, Sawczuk K, Herba E, Pojda-Wilczek D, Kostrzewa RM, Brus R. Nitric oxide modulates the amphetamine effect on [3H]glucose uptake in the brain of rats prenatally exposed to lead. Pharmacol Rep 2007;59(5):601-605.
|
| 85 | Bressler JP, Goldstein GW: Mechanisms of lead neurotoxicity. Biochem Pharmacol 1991;41(4):479-484.
https://doi.org/10.1016/0006-2952(91)90617-E |
| 86 | Kumawat KL, Kaushik DK, Goswami P, Basu A: Acute exposure to lead acetate activates microglia and induces subsequent bystander neuronal death via caspase-3 activation. Neurotoxicology 2014;41:143-153.
https://doi.org/10.1016/j.neuro.2014.02.002 |
| 87 | Malvezzi CK, Moreira EG, Vassilieff I, Vassilieff VS, Cordellini S. Effect of L-arginine, dimercaptosuccinic acid (DMSA) and the association of L-arginine and DMSA on tissue lead mobilization and blood pressure level in plumbism. Braz J Med Biol Res 2001;34(10):1341-1346.
https://doi.org/10.1590/S0100-879X2001001000016 |
| 88 | Nowak P, Szczerbak G, Nitka D, Kostrzewa RM, Sitkiewicz T, Brus R: Effect of prenatal lead exposure on nigrostriatal neurotransmission and hydroxyl radical formation in rat neostriatum: dopaminergic-nitrergic interaction. Toxicology 2008;246(1):83-89.
https://doi.org/10.1016/j.tox.2007.12.026 |
| 89 | Nowak P, Szczerbak G, Nitka D, Kostrzewa RM, Jośko J, Brus R: Cortical dopaminergic neurotransmission in rats intoxicated with lead during pregnancy. Nitric oxide and hydroxyl radicals formation involvement. Neurotoxicol Teratol 2008;30(5):428-432.
https://doi.org/10.1016/j.ntt.2008.02.003 |
| 90 | Sahin Z, Ozkaya A, Uckun M, Yologlu E, Kuzu M, Comakli V, Demirdag R, Tel AZ, Aymelek F, Yologlu S: Evaluation of the effects of Cyclotrichium niveum on brain acetylcholinesterase activity and oxidative stress in male rats orally exposed to lead acetate. Cell Mol Biol (Noisy-le-grand) 2019;65(5):3-8.
https://doi.org/10.14715/cmb/2019.65.5.2 |
| 91 | Brahma N, Gupta A: Acute toxicity of lead in fresh water bivalves Lamellidens jenkinsianus obesa and Parreysia (Parreysia) corrugata with evaluation of sublethal effects on acetylcholinesterase and catalase activity, lipid peroxidation, and behavior. Ecotoxicol Environ Saf 2020;189:109939.
https://doi.org/10.1016/j.ecoenv.2019.109939 |
| 92 | Zhang Y, Pei X, Jing L, Zhang Q, Zhao H. Lead induced cerebellar toxicology of developmental Japanese quail (Coturnix japonica) via oxidative stress-based Nrf2/Keap1 pathway inhibition and glutathione-mediated apoptosis signaling activation. Environ Pollut 2024;52:124114 .
https://doi.org/10.1016/j.envpol.2024.124114 |
| 93 | Liu CM, Zheng GH, Ming QL, Sun JM, Cheng C:. Protective effect of puerarin on lead-induced mouse cognitive impairment via altering activities of acetyl cholinesterase, monoamine oxidase and nitric oxide synthase. Environ Toxicol Pharmacol 2013;35(3):502-510.
https://doi.org/10.1016/j.etap.2013.02.009 |
| 94 | Phyu MP, Tangpong J: Neuroprotective effects of xanthone derivative of Garcinia mangostana against lead-induced acetylcholinesterase dysfunction and cognitive impairment. Food Chem Toxicol 2014;70:151-156.
https://doi.org/10.1016/j.fct.2014.04.035 |
| 95 | Liu CM, Ma JQ, Sun YZ: Puerarin protects rat kidney from lead-induced apoptosis by modulating the PI3K/Akt/eNOS pathway. Toxicol Appl Pharmacol 2012;258(3):330-342.
https://doi.org/10.1016/j.taap.2011.11.015 |
| 96 | Liu CM, Zheng GH, Ming QL, Sun JM, Cheng C: Protective effect of quercetin on lead-induced oxidative stress and endoplasmic reticulum stress in rat liver via the IRE1/JNK and PI3K/Akt pathway. Free Radic Res 2013;47(3):192-201.
https://doi.org/10.3109/10715762.2012.760198 |
| 97 | Liu CM, Zheng GH, Cheng C, Sun JM: Quercetin protects mouse brain against lead-induced neurotoxicity. J Agric Food Chem 2013;61(31):7630-7635.
https://doi.org/10.1021/jf303387d |
| 98 | Hegazy AA, Domouky AM, Akmal F, El-Wafaey DI. Possible role of selenium in ameliorating lead-induced neurotoxicity in the cerebrum of adult male rats: an experimental study. Sci Rep. 2023;13(1):15715.
https://doi.org/10.1038/s41598-023-42319-3 |
| 99 | Zeng PY, Tsai YH, Lee CL, Ma YK, Kuo TH. Minimal influence of estrous cycle on studies of female mouse behaviors. Front Mol Neurosci. 2023;16:1146109.
https://doi.org/10.3389/fnmol.2023.1146109 |
| 100 | Lovick TA, Zangrossi H Jr. Effect of Estrous Cycle on Behavior of Females in Rodent Tests of Anxiety. Front Psychiatry. 2021;12:711065.
https://doi.org/10.3389/fpsyt.2021.711065 |
| 101 | Zhang W, Lang R: Succinate metabolism: a promising therapeutic target for inflammation, ischemia/reperfusion injury and cancer. Front Cell Dev Biol 2023;11:1266973.
https://doi.org/10.3389/fcell.2023.1266973 |