Enhanced ISGylation via USP18 Isopeptidase Inactivation Fails to Mitigate the Inflammatory or Functional Course of Coxsackievirus B3-Induced Myocarditis
bDeutsches Zentrum für Herz-Kreislauf-Forschung, partner site Berlin, 10117 Berlin, Germany,
cUniversity of Freiburg, Institute of Neuropathology, 79106 Freiburg, Germany,
dCIBSS - Centre for Integrative Biological Signalling Studies, University of Freiburg, 79106 Freiburg, Germany,
eCardiopathology, Institute for Pathology and Neuropathology, University Hospital Tübingen, 72076 Tübingen, Germany
Keywords
Abstract
Background/Aims:
The ubiquitin-like protein ISG15 and its covalent conjugation to substrates (ISGylation) represent a critical interferon (IFN)-induced antiviral mechanism. USP18 is an ISG15-specific isopeptidase and a key negative regulator of type I IFN signaling. While inactivation of USP18’s catalytic activity enhances ISGylation and promotes viral resistance, its role in modulating inflammation and cardiac function during CVB3-induced myocarditis remains unclear. This study aimed to determine whether selective inactivation of USP18 isopeptidase activity influences the inflammatory and functional course of viral myocarditis.Methods:
Usp18 C61A/C61A knock-in mice, which lack USP18 isopeptidase activity but retain IFN regulatory function, were used on both C57BL/6 and A/J backgrounds. Mice were infected with the cardiotropic CVB3-Nancy strain, and disease progression was assessed through virological, histological, immunological, and echocardiographic analyses. Immune cell infiltration was quantified by flow cytometry, and ISGylation was assessed by immunoblotting.Results:
Despite enhanced ISGylation, Usp18 C61A/C61A mice did not exhibit altered cardiac viral titers or inflammation compared to wild-type controls. Histological scores and immune cell composition in the heart were similar between genotypes in both C57BL/6 and A/J backgrounds. Echocardiography confirmed functional impairment following CVB3 infection but revealed no significant genotype-dependent differences in cardiac performance. Inflammatory cytokine expression was largely unaffected by enhanced ISGylation, with only minor differences observed.Conclusion:
While ISGylation is critical for antiviral protection in CVB3 infection, selective inactivation of USP18 isopeptidase activity does not mitigate myocardial inflammation or dysfunction during established CVB3 myocarditis. These findings suggest that therapeutic enhancement of ISGylation alone may be insufficient to control inflammation-driven cardiac damage in this model.Introduction
Coxsackievirus B3 (CVB3), a member of the Enterovirus genus within the Picornaviridae family, is a small, non-enveloped virus with a single-stranded, positive-sense RNA genome and a prominent etiological agent of viral myocarditis [1, 2]. Following fecal-oral transmission, CVB3 initially replicates in the gastrointestinal tract before disseminating via viremia to secondary target organs such as pancreas, liver, and notably, the heart [3-5]. In a subset of patients, especially children and young adults, CVB3 infection leads to acute myocarditis, and approximately 10–20% of these cases may progress to chronic dilated cardiomyopathy (DCM), a frequent indication for heart transplantation [2, 6, 7].
Due to the rarity of early-stage human biopsies, mechanistic insights into CVB3-induced myocarditis primarily derive from murine models. In susceptible mouse strains, CVB3 shows high tropism for cardiac tissue with peak viral replication around day 4–6 post-infection, followed by immune-mediated tissue damage manifesting as acute myocarditis [8, 9]. Viral cytopathogenicity is mediated by proteases such as 2A and 3C, which cleave host proteins like eIF4G and dystrophin, suppressing host translation and compromising cardiomyocyte integrity [10-12]. In parallel, the host initiates a robust innate immune response. Recognition of viral RNA by pattern recognition receptors (PRRs) such as RIG-I, MDA5, TLR3 and TLR7/8 leads to induction of type I interferons (IFNs) and inflammatory mediators [13-16]. CVB3 proteases can antagonize these pathways by targeting key PRR components [17, 18], which underscores the essential role of intact type I IFN signaling for controlling early viral replication and reducing disease severity [19-21].
One of the key effector pathways downstream of type I IFN signaling is the ISG15 system. ISG15 is a ubiquitin-like protein induced by IFN signaling and expressed as a 17 kDa precursor that is processed into a 15 kDa mature form [22, 23]. It can exist intracellularly in unconjugated form, function in a cytokine-like manner extracellularly, or be covalently attached to substrate proteins via ISGylation [24-26]. The ISGylation cascade is mediated by the E1 enzyme Ube1L, E2 enzyme Ube2L6, and E3 ligases such as Herc6 in mice [26-29]. This modification alters the function, stability, and interactions of target proteins-including other key components of the interferon response, such as IRF3, IFITs and RIG-I [9, 30, 31].
In the context of CVB3-induced myocarditis, the ISG15 system plays a central antiviral and immunoregulatory role. Mice deficient in ISG15 or its E1-conjugating enzyme Ube1L exhibit significantly elevated viral titers in the heart, enhanced inflammation, cardiac dysfunction, and increased mortality, indicating that protein ISGylation is indispensable for antiviral defense [9, 32]. One putative mechanism involves the ISGylation of the viral 2A protease, which reduces its ability to cleave eIF4G, thereby protecting host protein synthesis during infection [32]. Recent data further highlight that ISGylation synergistically enhances the antiviral function of other IFN-stimulated proteins, particularly IFIT1 and IFIT3, and is essential to establish resistance in non-hematopoietic cells, such as cardiomyocytes. In CVB3 infection, ISGylation not only restricts viral replication but also modulates liver energy metabolism by promoting oxidative phosphorylation, thus supporting systemic adaptation to infection-induced stress [9]. Complementary findings in cardiac tissue have shown that ISGylation affects also cardiac metabolism [33]. In CVB3-infected mice, ISG15 deficiency results in cardiac atrophy and reduced cardiac output. Proteomic analysis identified key glycolytic enzymes - hexokinase 2 and phosphofructokinase 1 - as major ISGylation substrates in the infected heart. Functional studies demonstrated that ISGylation suppresses their enzymatic activity, thereby preventing the IFN-induced glycolytic shift commonly observed during infection. Instead, the ISG15 system preserves oxidative metabolism, supports higher ATP production capacity, and protects against energy failure in cardiomyocytes [33]. These findings underscore that ISG15 safeguards energy homeostasis under infectious stress, contributing to functional preservation of the heart.
The ISG15-specific isopeptidase USP18 is a crucial negative regulator of this system. USP18 removes ISG15 from substrates and simultaneously attenuates type I IFN signaling by interfering with JAK1 recruitment to the IFN receptor IFNAR2 [34, 35]. While Usp18 -deficient mice exhibit severe IFN hypersensitivity and developmental abnormalities due to loss of non-catalytic functions [34, 36], selective inactivation of its isopeptidase activity in Usp18 C61A/C61A knock-in mice leads to increased ISGylation without affecting viability or baseline IFN regulation [37]. Importantly, these mice show enhanced resistance to viral infections, providing evidence that stabilizing ISGylation by inactivating USP18’s protease activity can boost antiviral effector function without triggering deleterious immune overactivation [37]. Strikingly, in cells with abrogated deISGylating enzymatic activity, increased ISGylation renders cells more resistant to various CV strains, suggesting that pharmacological stabilization of ISGylation by USP18 protease inhibition could be therapeutically exploited [9, 38]. Beyond this receptor-level and enzymatic regulation, emerging evidence points to additional layers of USP18 function that act independently of IFNAR interaction. USP18 also modulates transcription more broadly: Arimoto et al. demonstrated that nuclear USP18, in cooperation with NFκB, diminishes binding of IFN-regulated transcription factors to their corresponding DNA [39]. This function allows USP18 to regulate both typical ISGs and non-canonical ISGs that are important for processes such as cancer cell pyroptosis [39]. Moreover, USP18 was shown to control cell polarization in tumor-associated macrophages. Specifically, USP18 stabilizes the colony stimulating factor 1 receptor (CSF1R), thereby preventing its degradation and skewing macrophages toward an anti-inflammatory phenotype, with significant implications for the composition of the tumor microenvironment [40]. Together with findings by Fan et al. (2020), who identified ISGylation as a key mechanism enabling STAT1/2 clustering and chemokine gene activation in antitumor immunity [41], these studies highlight USP18 as a multifaceted immune regulator - balancing enzymatic ISGylation control, receptor-level IFN modulation, and nuclear transcriptional repression of immune programs. These complex regulatory functions of USP18 raise the question to what extent its enzymatic activity contributes to antiviral protection and immune regulation in the heart during enteroviral infection.
Taken together, CVB3-induced myocarditis results from a complex interaction between viral cytotoxicity and host immune defenses. The ISG15 system has emerged as a central player in this context, acting downstream of type I IFN signaling to provide direct antiviral protection and to preserve cardiac metabolic homeostasis through protein ISGylation. Functional studies in ISG15-deficient models have established the critical importance of this post-translational modification in cardiomyocytes for viral containment and tissue integrity [9, 32, 33]. At the same time, USP18 has been identified as a multifaceted regulator of inflammation, integrating enzymatic control of ISGylation with broader roles in IFNAR signaling and transcriptional repression of both canonical and non-canonical ISGs in various immune and non-immune cells. Among these roles, its specific function as an ISG15 isopeptidase remains an attractive therapeutic target, as selective inactivation of its enzymatic activity enhances ISGylation and antiviral resistance without triggering IFN-related toxicity [37]. This study aims to dissect the contribution of USP18’s deISGylating activity to the course of CVB3 myocarditis. By employing Usp18 C61A/C61A knock-in mice, we investigate whether increased ISGylation alters the severity of myocardial inflammation and preserves cardiac function. This targeted approach enables mechanistic insight into how enzymatic regulation of ISGylation by USP18 affects the inflammatory and functional outcome of viral heart disease and evaluates the therapeutic potential of selectively modulating USP18 activity in a preclinical mammalian model system.
Materials and Methods
Animals
The generation and basic characterization of Usp18 C61A/C61A knock-in mice, which lack
USP18 isopeptidase activity due to a cysteine-to-alanine substitution in the catalytic core, has been
described elsewhere [37]. These knock-in mice, originally generated on C57BL/6 background (entitled
Usp18 C61A/C61A - B6), were backcrossed with A/J mice for five generations to generate
Usp18 C61A/C61A mice on a mixed A/J X C57BL/6 background (entitled Usp18
C61A/C61A - A/J). All mice were bred in the internal breeding facility of Charité
– Universitätsmedizin Berlin. Four-week-old, male Usp18 +/+-B6 and Usp18
C61A/C61A - B6 and seven-week-old, male Usp18 +/+ - A/J and Usp18
C61A/C61A - A/J mice, both originating from Usp18 +/C61A X Usp18
+/C61A breeding in the respective background, were infected intraperitoneally with a single
injection of Coxsackievirus B3 (CVB3) Nancy strain [42]. The infection dose was 1 × 10⁵
plaque-forming units (PFU) for Usp18 C61A/C61A - B6 mice and 1 × 10⁴ PFU for
Usp18 C61A/C61A - A/J mice. Wild-type littermates of both strains were infected
following the same protocol and served as controls. Body weight was recorded daily. Rectal body
temperature and tail vein blood glucose levels (Accu-Chek glucometer, Roche Diabetes Care, Indianapolis,
IN, USA) were measured during echocardiography at baseline and on day 7 post infection in Usp18
C61A/C61A - A/J mice. The experiments were terminated eight days post-infection for
Usp18 C61A/C61A - B6 mice and seven days post-infection for Usp18 C61A/C61A
- A/J mice. Tissue collection in mice on A/J background was performed on day 7 post-infection due to
the pronounced systemic phenotype associated with CVB3-Nancy infection in this model, including
significant weight loss. To avoid exceeding humane endpoints and ensure compliance with ethical
guidelines, animals were sacrificed at this earlier time point. To examine the kinetics of chemokine and
cytokine gene expression, Usp18 C61A/C61A - B6 mice were also sacrificed at day 0
(uninfected), day 1.5, day 3, and day 6 post-infection. Mice were euthanized by a lethal overdose of
isoflurane (CP-Pharma, Burgdorf, Germany). Hearts were perfused transmurally with cold PBS, and
subsequently heart, spleen, liver, and pancreas were collected. Tissue samples were snap-frozen in liquid
nitrogen and stored at −80°C for later analysis. Samples for histology were fixed in 4%
formaldehyde (Carl Roth, Karlsruhe, Germany) and embedded in paraffin.
All animal experiments were conducted in accordance with the “Guide for the Care and Use of
Laboratory Animals”, the German Animal Welfare Act, and European Directive 2010/63/EU on the
protection of animals used for scientific purposes. The study was approved by the local Animal Welfare
Authority in Berlin (“Landesamt für Gesundheit und Soziales”) and registered under the
approval numbers H0076/08, G0119/20, G0272/14, G0279/11. To minimize animal suffering, tramadol
(Grünenthal, Stolberg, Germany) was administered orally via drinking water to all Usp18
C61A/C61A - A/J mice, following a previously published protocol [43]. Mice meeting the
predefined termination criteria outlined in the score sheets were euthanized prematurely.
Histology
Paraffin-embedded heart and pancreas samples were stained with hematoxylin and eosin (H&E) and
evaluated by a blinded pathologist. Cardiac inflammation was assessed using an established scoring system
(0: no inflammatory infiltrates, 1: small foci of inflammatory cells between myocytes, 2: larger foci with
>100 inflammatory cells, 3: ≤10% of the cross-section involved, 4: 10%–30% of the
cross-section involved) [44]. Pancreatic injury was quantified as the percentage of tissue destruction,
ranging from 0% to 100%.
Echocardiography
Echocardiography was performed on Usp18 C61A/C61A - A/J mice 1 or 2 days before
infection (baseline) and before euthanasia on day 7 post-infection. The echocardiographic setup included a
Vevo3100 ultrasound system (FUJIFILM VisualSonics, Toronto, ON, Canada), an MX400 high-resolution
ultrasound probe, a heated examination table with built-in electrocardiogram (ECG) recording, a pre-warmed
ultrasound gel (Parker Laboratories, Fairfield, NJ, USA) and a heat lamp. Anesthesia of the mice was
induced with approximately 3% isoflurane (CP-Pharma) and maintained at a concentration of 2% throughout
the procedure. Heart rate, respiratory rate and body temperature were continuously monitored and
maintained within physiological ranges. After the mice were shaved on the left lateral thorax,
echocardiographic images were acquired from standard views according to published guidelines [45].
Briefly, B-mode images were obtained from the parasternal long-axis view, parasternal mid-papillary
short-axis view and 4-chamber view. Additionally, M-mode images were recorded in the mid-papillary
short-axis view, while mitral valve pulsed-wave (PW) Doppler and septal mitral anulus tissue Doppler
measurements were performed in the 4-chamber view. The acquired images were stored and later analyzed
using VevoLAB 3.2.1 software (FUJIFILM VisualSonics).
Each echocardiographic parameter was derived from the average of at least three recorded images. The
endomyocardial border in long-axis view images was traced using the AutoLV tool and Simpson method was
applied to measure left ventricular end-diastolic volume, end-systolic volume, stroke volume, cardiac
output and ejection fraction. AutoLV analysis of mid-papillary short-axis M-mode images provided
measurements of left ventricular anterior and posterior wall thickness as well as left ventricular
internal diameter in diastole. Left ventricular mass was calculated. Mitral valve early (MV E) and atrial
(MV A) diastolic waves, isovolumetric relaxation (IVRT) and contraction (IVCT) time, aortic ejection time
(AET), mitral valve E deceleration time and mitral anulus early diastolic (MV E’), atrial (MV
A’) and systolic (MV S’) waves were measured in the mitral valve PW Doppler and septal mitral
anulus tissue doppler images, respectively. The myocardial performance index (Tei-index) and the ratios MV
E/A, E’/A’ and E/E’ were calculated.
Immune cell isolation from heart tissue
Excised heart tissue was first weighed and then finely minced in cold RPMI medium (Life Technologies,
Waltham, MA, USA), which was supplemented with 2% fetal calf serum (FCS; Sigma-Aldrich, St. Louis, MO,
USA), 30 mM HEPES (Life Technologies), and 1% penicillin/streptomycin (Life Technologies). To facilitate
enzymatic digestion, collagenase Type II (Worthington, Lakewood, NJ, USA) and DNase I (Sigma-Aldrich) were
added at final concentrations of 1 mg/ml and 0.15 mg/ml, respectively. The tissue suspension was then
incubated at 37°C for 30 minutes with continuous stirring. 10 mM EDTA (VWR) was added to stop
enzymatic activity and subsequently the digested tissue was passed through a 70 µm cell strainer
(Corning, Corning, NJ, USA) to remove undigested fragments. The filtrate was then centrifuged at 310
× g for 10 minutes at 4 °C, after which the supernatant was discarded. To lyse red blood cells,
the pellet was resuspended in an erythrocyte lysis buffer (ACK buffer) consisting of 10 mM KHCO₃
(Carl Roth), 155 mM NH₄Cl (Thermo Fisher Scientific, Waltham, MA, USA), and 0.1 mM EDTA (VWR), and
incubated at room temperature for 3 minutes. Following an additional centrifugation step, the remaining
leukocyte pellet was resuspended in FACS buffer supplemented with 2% FCS (Sigma-Aldrich) and 2 mM EDTA for
downstream analysis.
Flow cytometry
Immune cells extracted from a defined amount of heart tissue (15 or 20 mg) were processed for further flow
cytometry staining. To minimize nonspecific antibody binding, cells were first incubated with an Fc
blocking reagent (Miltenyi) at a 1:50 dilution for 20 minutes at 4 °C. This was followed by staining
with a pre-mixed antibody-fluorochrome cocktail for another 20 minutes at 4 °C in the dark. The
antibody-fluorochrome conjugates were purchased from BD Bioscience (Heidelberg, Germany), BioLegend (San
Diego, CA, USA), and Life Technologies, with further details available in Supplemental Table S1. After
staining, cells were washed with PBS, centrifuged and subjected to cell viability staining using fixable
viability dye eFluor 780 (eBioscience, San Diego, CA, USA) at a 1:1000 dilution in PBS for 30 minutes in
the dark. The samples were then washed, fixed in 2% formaldehyde (Carl Roth), washed again and resuspended
in FACS buffer. To determine the absolute number of immune cells, 123 count eBeads (Life Technologies)
were added to the samples. Flow cytometry was performed using either a FACSymphony (BD Bioscience) or
LSRII (BD Bioscience) flow cytometer. Data analysis was carried out using FlowJo V10.6.2 software. The
spectral overlap of the fluorochromes was compensated using single stains. Representative gating
strategies are shown in Supplementary Figures S1-S4. Fluorescence minus one (FMO) controls were used to
assist in the selection of accurate gates.
Quantification of viral titer
To determine viral titers in tissue homogenates from infected C57BL/6 mice, plaque assays were performed
using confluent GMK cell monolayers. Serial ten-fold dilutions of virus-containing samples were prepared
in serum-free DMEM and applied to the cell layers. Plates were incubated for 1 hour at 37°C with 5%
CO₂, with gentle agitation every 10–15 minutes. The inoculum was then removed, and cells were
overlaid with DMEM containing 0.6% agarose, 2.5% fetal calf serum (FCS), 1% penicillin/streptomycin, 1 mM
sodium pyruvate, and 3.75% sodium bicarbonate (pH 7.4). After 48 hours of incubation at 37°C, cells
were fixed with a methanol/acetic acid solution and stained with 0.25% crystal violet to visualize
plaques. For infected A/J mouse tissues, viral titers were quantified by plaque assays on confluent HeLa
cell monolayers. Serial ten-fold dilutions of virus-containing samples were prepared in PBS and added to
the cell layers. After a 30-minute incubation at 37°C with 5% CO₂, the inoculum was removed,
and cells were overlaid with Eagle’s agar composed of MEM, 0.68% penicillin/streptomycin, 1.6 g/L
sodium bicarbonate (Carl Roth), 9% FCS, and 0.7% Difco Agar Noble (BD Bioscience). Plates were incubated
at 37°C for 48 hours. Plaques were visualized by staining with MTT (3-(4, 5-dimethylthiazol-2-yl)-2,
5-diphenyltetrazolium bromide; Sigma-Aldrich) for 1 hour at room temperature. Plaques were counted, and
viral titers were calculated as plaque-forming units (PFU) per gram of tissue.
RNA Isolation and Quantitative Real-Time PCR (qPCR)
RNA extraction from organ samples was performed using the TRIzol reagent (Thermo Fisher Scientific)
according to the manufacturer’s protocol. Following tissue homogenization in TRIzol, RNA was
isolated via phase separation with chloroform, precipitated with isopropanol, washed with 75% ethanol, and
subsequently dissolved in DEPC-treated water. RNA quantity was assessed by NanoDrop spectrophotometer
(VWR, Radnor, PA/USA). An RNA amount ranging from 250 to 1000 ng was converted into cDNA using MLV Reverse
Transcriptase (Promega, Madison, WI, USA) in combination with random hexamer primers (Roche). For the
relative quantification of Il-1β (interleukin-1β) , Il-6 (interleukin-6) , Tnf-
α (tumor necrosis factor- α ), Ccl2 and Cxcl2 mRNA expression,
primers and probes of TaqMan gene expression assays (Thermo Fisher Scientific) were used. Cvb3
genome and Hprt (hypoxanthinguanin -phosphoribosyltransferase ) gene expression were
quantified using the following combinations of primers and probes: Hprt forward: 5ʹ-ATC ATT
ATG CCG AGG ATT TGG AA-3ʹ, reverse: 5ʹ-TTG AGC ACA CAG AGG GCC A-3ʹ, probe: 5ʹFAM- TGG
ACA GGA CTG AAA GAC TTG CTC GAG ATG-3ʹTAMRA; Cvb3 forward: 5′-CCC TGA ATG CGG CTA ATC
C-3, reverse: 5′-ATT GTC ACC ATA AGC AGC CA-3′, probe: 5′-FAM-TGC AGC GGA ACC G-MGB3.
The reactions were performed on the StepOnePlus real-time PCR system (Thermo Fisher Scientific), with gene
expression normalized to the housekeeping gene Hprt and calculated as ΔCt values.
Protein Quantification and Immunoblotting
Snap-frozen heart tissue was added to Lysis Matrix D tubes with RIPA-buffer with 1, 25X Protease Inhibitor
Cocktail (Roche) and tissue was homogenized 3 times for 30 seconds using the MP Biomedical FastPrep,
centrifuged and the supernatant was used for further analysis. Protein concentration was determined using
the Bradford assay (Thermo Fisher Scientific) following the manufacturer’s instructions. Tissue
lysates were denatured, and the same protein amount was loaded onto an 8%-SDS-PAGE gel for protein
separation followed by transfer to nitrocellulose membranes (Odyssey Nitrocellulose Membranes, LI-COR
Biotechnology, Lincoln, NE, USA). The membranes were blocked for 1h at room temperature in Rotiblock and
incubated with the primary antibody against mISG15 (lab stock provided by KP Knobeloch, Freiburg, Germany,
dilution 1:1, 000 in Rotiblock) at 4°C overnight, washed with PBS-T, and incubated with goat
anti-rabbit immunoglobulin IRDyeTM 800CW (LI-COR, dilution 1:10, 000 in PBS-T) for 1h at room temperature,
washed, imaged and then incubated with the primary antibody against mTubulin (Merck, Sigma Aldrich,
dilution 1:5, 000 in Rotiblock, clone DM1A), washed, and incubated with goat anti-mouse immunoglobulin
IRDyeTM 680LT (LI-COR, dilution 1:20, 000 in PBS-T) and imaged. Protein detection was performed using the
Odyssey CLx infrared imaging system (LI-COR Biotechnology) and the analysis was carried out using Image
Studio Lite software version 5.2 (LI-COR Biotechnology). Western blot results were quantified using
densitometry. After correcting for background values, ISGylation smear signal was measured and normalized
to Tubulin as loading control.
Statistics
Statistical analyses were conducted using GraphPad Prism v10.00 for Windows (La Jolla, CA, USA). Outliers
in the primary data were detected using the ROUT method (Q = 1%) and removed from further analysis. If not
otherwise indicated, data are presented as individual data points, with results expressed as mean ±
SEM. Viral titer data were logarithmically transformed prior to graphical representation and statistical
analysis. As part of the statistical tests, the normality of the data distribution was first assessed
using the D’Agostino-Pearson test. For comparisons between two groups with equal variance, unpaired
t -tests were performed (Figures 1D, 2A-E, 2G, 3B, 3H, 4A-B, 4D-F, 4H). If the variance between two
groups was unequal, an unpaired Welch´s t -test was performed (1E, 3E, 3G). Datasets
containing the two factors “time after infection” and “genotype” were
statistically analyzed with 2-way ANOVA analysis (2H, 5A-F) or repeated measures mixed-effect models
(Figures 1B, 3C-D). Subsequently, post-hoc tests were applied for multiple comparisons. The specific
statistical tests used are detailed in the figure legends for each graph. The significance threshold was
set to p < 0.05 in all used tests. Significant results (p < 0.05) are marked with asterisks (*),
non-significant results are not indicated as such.
Results
Inactivation of USP18 isopeptidase activity is associated with unchanged levels of CVB3-induced
myocardial inflammation in C57BL/6 mice
In Usp18 C61A/C61A mice, a catalytically inactive variant of USP18 is
expressed that lacks deISGylating activity, but retains its negative regulatory function on IFN signaling.
As a result, ISGylation induced by CVB3 infection is selectively stabilized, which has recently been shown
to suppress viral replication in both CVB3 infected cardiomyocytes and mouse hearts [9]. To investigate
the effects attributed to preserved ISGylation during the acute phase of myocarditis, we utilized the same
experimental setup as recently and analyzed mice on day 8 post-infection, which is consistent with the
peak inflammatory response in murine heart tissue [32, 46]. Wild-type (Usp18 +/+ - B6)
and Usp18 C61A/C61A mice (Usp18 C61A/C61A - B6), both on a C57BL/6
background, were infected intraperitoneally with 1×10⁵ PFU Coxsackievirus B3 (CVB3), monitored
throughout the infection, and sacrificed on day 8 post-infection (p.i.) for analysis of key parameters
(Fig. 1A). Body weight declined significantly over the course of infection, with no significant
differences observed between Usp18 +/+ and Usp18 C61A/C61A - B6
mice (Fig. 1B). The pancreas, a highly susceptible organ during CVB3 infection, exhibited extensive
tissue destruction in all animals, irrespective of genotype (Fig. 1C). Previous studies demonstrated that
protein ISGylation exerts antiviral effects in the hearts at day 6 post-infection, as evidenced by reduced
viral titers and lower CVB3 RNA levels in Usp18 C61A/C61A - B6 mice [9]. To determine
whether this protective effect is sustained during the peak of myocardial inflammation, we analyzed viral
loads and CVB3 RNA expression in cardiac tissue at day 8 post-infection. All mice exhibited successful
infection, as indicated by consistent pancreatic tissue injury across experimental groups (Fig. 1C). In
line with previous findings showing limited antiviral activity of protein ISGylation at later stages of
infection [9], neither viral titers nor CVB3 RNA levels significantly differed between Usp18
+/+ and Usp18 C61A/C61A - B6 mice (Fig. 1D-E). Likewise, the antiviral
effect observed at day 6 was no longer evident at day 8, with no significant differences in viral titers,
or CVB3 RNA levels between hearts of Usp18 +/+ and Usp18 C61A/C61A -
B6 mice.
To determine whether enhanced ISGylation in Usp18 C61A/C61A – B6 mice [9]
influence inflammation and immune cell infiltration in the heart, myocarditis severity was assessed by
standardized histological scoring 8 days after infection, when immune cell infiltration peaks. Upon CVB3
infection, all animals exhibited cardiac inflammation and immune cell infiltration. Of note, some mice
developed large foci containing >100 inflammatory cells, further validating the robustness of this
myocarditis model (Fig. 2A). However, no significant differences in myocarditis scores were observed
between Usp18 +/+ and Usp18 C61A/C61A - B6 mice at this stage. To
further characterize and quantify immune cell infiltration in the heart, flow cytometry was performed
(gating strategy shown in Supplementary Figures S1, S2 and S4). Profound CD45⁺ immune cell
infiltration was evident post-infection, but no significant differences were detected between Usp18
+/+ and Usp18 C61A/C61A - B6 mice (Fig. 2B). Within the myeloid
compartment, no significant differences were found in the frequencies of CD11b⁺Ly6G⁺
neutrophils, CD11b⁺F4/80⁻ monocytes or CD11b⁺F4/80⁺ macrophages between Usp18
+/+ and Usp18 C61A/C61A - B6 animals (Fig. 2C). Usp18
C61A/C61A - B6 mice exhibited a significantly higher abundance of CD11b⁺CD11c⁺
dendritic cells in the heart following CVB3 infection (Fig. 2C). Within the lymphoid compartment, no
significant differences were observed in CD19+B220⁺ B cells, CD3⁺CD4⁺ T cells
or CD3⁺CD8⁺ T cells between Usp18 +/+ and Usp18 C61A/C61A
- B6 mice (Fig. 2D). Within the monocyte compartment, we analyzed the percentage of
Ly6Chigh inflammatory monocytes, well-known regulators of CVB3-induced myocardial inflammation
[46-49], and observed no differences between genotypes (Fig. 2E-F). Also, a subset of dendritic cells
expressed Ly6C, suggesting they are monocyte-derived dendritic cells, a population known to expand under
inflammatory conditions [50, 51] (Fig. 2G). To further assess the inflammatory response over time, we
measured mRNA expression levels of key inflammatory cytokines and chemokines in heart tissue at baseline
and 1.5, 3, 6 and 8 days post-infection. Tnf- α , Il-1 β , Il-6
, Ccl2 , and Cxcl2 expression peaked at day 3 p.i. and gradually declined thereafter
(Fig. 2H). Notably, enhanced ISGylation in Usp18 C61A/C61A - B6 mice did not enhance
cytokine or chemokine expression. In contrast, Cxcl2 expression was reduced in Usp18
C61A/C61A - B6 mice 3 days p.i., while Tnf- α , Il-1 β
, Il-6 and Ccl2 levels remained unaltered. Together, these data indicate that enhanced
ISGylation resulting from selective inactivation of USP18 isopeptidase activity in C57BL/6 mice does not
markedly alter cardiac inflammation, immune cell composition, or cytokine responses during CVB3-induced
myocarditis.
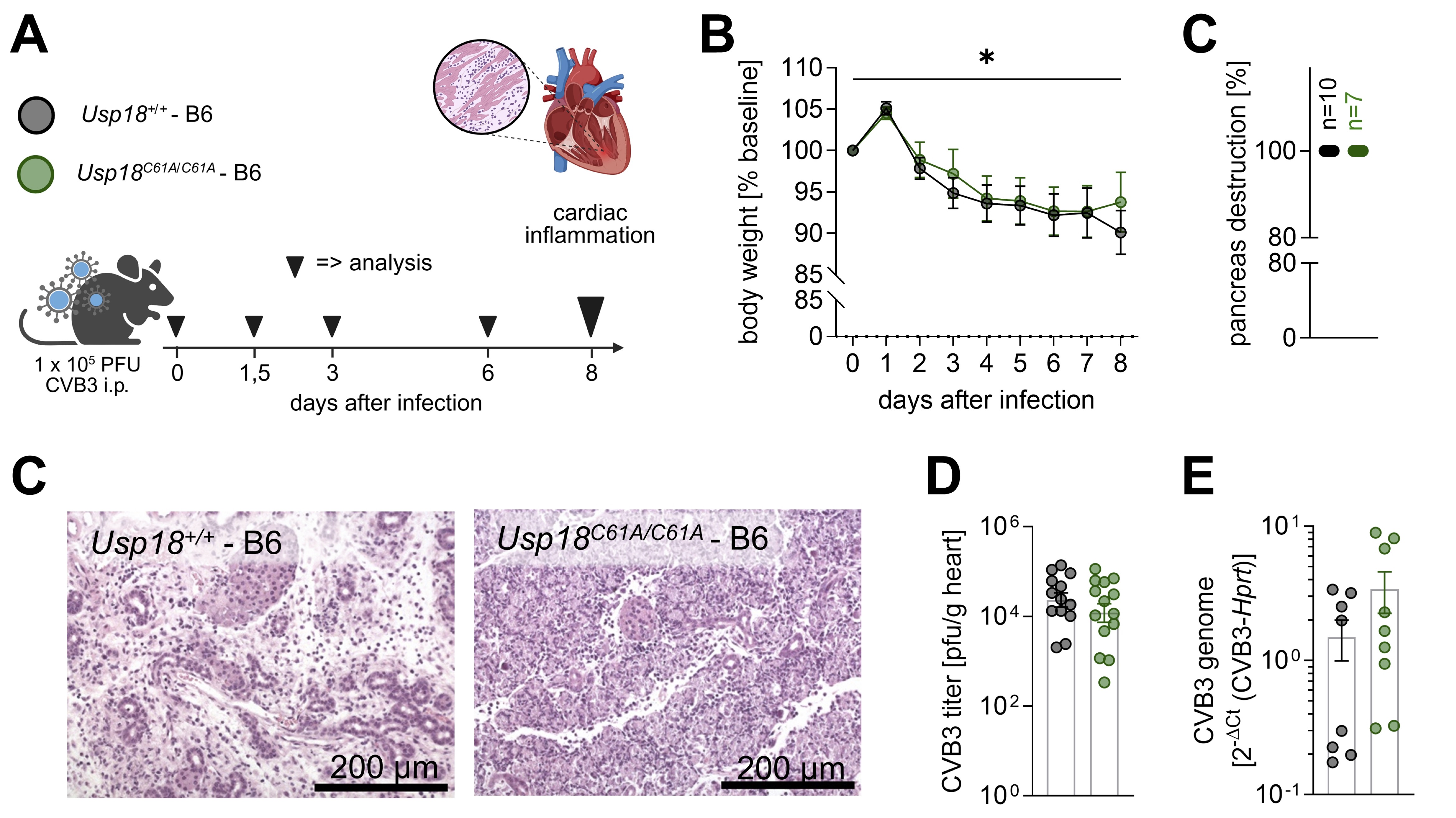
Fig. 1: Inactivation of USP18 isopeptidase activity in Usp18C61A/C61A - B6 mice does not alter systemic responses in CVB3 infection. A: Schematic of the experimental setup used to study the effects of enhanced ISGylation on cardiac inflammation during CVB3 infection. Four-week-old male Usp18C61A/C61A - B6 mice and their Usp18+/+ controls were intraperitoneally infected with 1x 105 PFU CVB3. In addition to the main analysis time point at day 8 (nC61A/C61A = 22, n+/+ = 20; large triangle) post-infection, which focuses on key markers of cardiac inflammation, mice were sacrificed at day 0 (uninfected; nC61A/C61A = 3, n+/+ = 3), day 1.5 (nC61A/C61A = 4, n+/+ = 4), day 3 (nC61A/C61A = 4, n+/+ = 4), and day 6 (nC61A/C61A = 4, n+/+ = 4) post infection to examine the kinetics of chemokine and cytokine gene expression (small triangle). B: Body weight was measured daily during infection and was normalised to baseline body weight. The data was statistically analysed using 2-way repeated measures ANOVA (mixed effects model). All time points were compared to baseline regardless of genotype using Dunnett post-hoc test. For clarity, only the results for the comparison baseline – day 8 are indicated in the graphs. Sidak post-hoc tests revealed no significant differences between genotypes at any time point. C: The degree of pancreatic destruction in haematoxylin and eosin (H&E)-stained sections was graded (nC61A/C61A = 7, n+/+ = 10) and representative images are shown for both genotypes. The corresponding scale bar (200 µm) is indicated. D-E: Viral load in heart tissue was determined by (D) plaque assay (nC61A/C61A = 13; n+/+ = 14) and (E) TaqMan qPCR (nC61A/C61A = 9; n+/+ = 8). The plaque assay data were logarithmically transformed before plotting. Statistical testing was performed using an unpaired t-test and Welch´s t-test, respectively. Significant results (p < 0.05) are marked with an asterisk (*), non-significant results are not indicated as such.
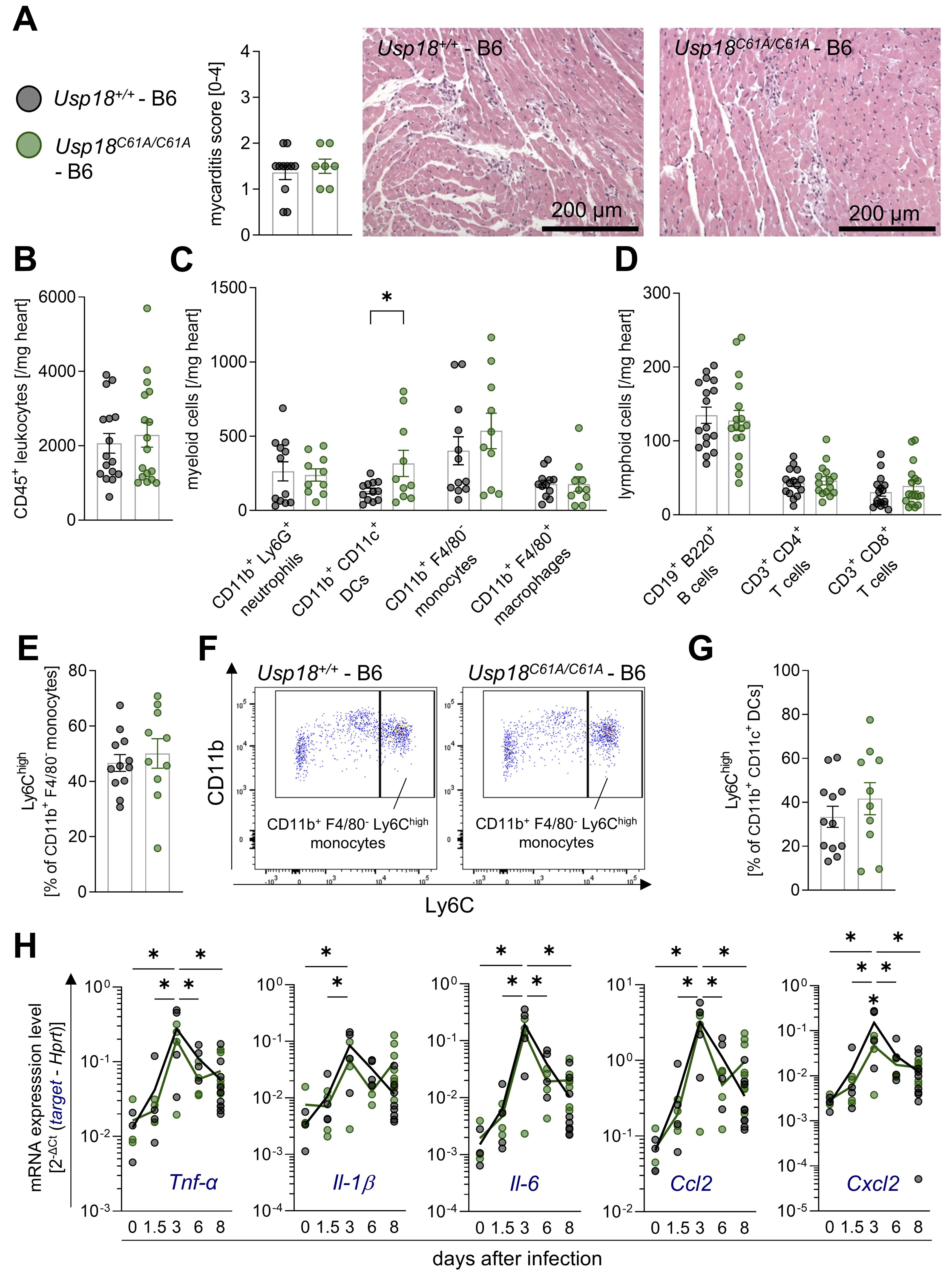
Fig. 2: Despite enhanced ISGylation, Usp18C61A/C61A - B6 mice exhibit sustained cardiac inflammation during CVB3 infection. Four-week-old male Usp18C61A/C61A - B6 mice (nC61A/C61A = 22) and their Usp18+/+ - B6 control mice (n+/+ = 20) were intraperitoneally infected with 1 x 105 PFU CVB3 and analyzed at day 8 post infection. Additionally, mice were sacrificed at day 0 (uninfected; nC61A/C61A = 3, n+/+ = 3), day 1.5 (nC61A/C61A = 4, n+/+ = 4), day 3 (nC61A/C61A = 4, n+/+ = 4), and day 6 (nC61A/C61A = 4, n+/+ = 4) post-infection to examine the kinetics of chemokine and cytokine gene expression. A: Heart tissue samples were stained with haematoxylin and eosin (H&E) and the myocarditis score was assessed (nC61A/C61A = 7, n+/+ = 11). An unpaired t-test showed no statistically significant differences. Representative images are shown for both genotypes. The corresponding scale bar (200 µm) is indicated. B: CD45+ leukocyte count per milligram cardiac tissue was measured by flow cytometry (nC61A/C61A = 17, n+/+ = 16). C: The cell count per milligram cardiac tissue was determined by flow cytometry for the following myeloid subpopulations: CD11b+ Ly6G+ neutrophils, CD11b+ CD11c+ dendritic cells (DCs), CD11b+ F4/80- monocytes and CD11b+ F4/80+ macrophages (nC61A/C61A = 10, n+/+ = 12). D: The cell count per milligram cardiac tissue was determined by flow cytometry for the following lymphoid subpopulations: CD19+ B220+ cells, CD3+ CD4+ T cells and CD3+ CD8+ T cells (nC61A/C61A = 17, n+/+ = 17). E: The percentage of CD11b+ F4/80- monocytes highly expressing Ly6C (Ly6Chigh) is shown (nC61A/C61A = 10, n+/+ = 12). F: Representative gating plots of CD11b+ F4/80- Ly6Chigh monocytes are depicted for both genotypes. G: The percentage of CD11b+ CD11c+ DCs highly expressing Ly6C (Ly6Chigh) is shown (nC61A/C61A = 10, n+/+ = 12). Data shown in graph 2B – 2G were statistically analyzed by unpaired t-tests, in which each immune cell population was tested separately without correction for multiple comparison. H: Chemokine and cytokine gene expression was analyzed in cardiac tissue from mice sacrificed on day 0 (uninfected), day 1.5, day 3, day 6 and day 8 (nC61A/C61A = 9; n+/+ = 10), using the following target genes: tumor necrosis factor α (Tnf-α), interleukin-1b (Il-1), interleukin-6 (Il-6) and the chemokines Ccl2 and Cxcl2. Hypoxanthine-guanine phosphoribosyltransferase (Hprt) was used as a housekeeping gene. Results are presented as 2-ΔCt (target - Hprt). Data was statistically analysed with a 2-way ANOVA. Sidak multiple comparison of each time points against all other time points regardless of the genotype resulted in statistically significant results, which are indicated by asterisks and black lines. In addition, differences between genotypes were analyzed using Sidak multiple comparisons. Significant results (p < 0.05) are indicated by single asterisks above the corresponding time point. Non-significant results are not indicated as such.
Viral load and cardiac inflammation remain unaffected in Usp18 C61A/C61A - A/J mice despite
elevated ISGylation
To further investigate the potential anti-inflammatory effects of enhanced ISGylation in a setting of more
pronounced myocardial pathology, we transitioned from the relatively resistant C57BL/6 background to the
more susceptible A/J mouse strain. While C57BL/6 mice infected with the cardiotropic CVB3-Nancy strain
develop mild acute myocarditis, A/J mice exhibit significantly greater susceptibility to infection [46,
52]. To evaluate whether stabilization of protein ISGylation through USP18 isopeptidase inactivation
modulates the extent of myocardial inflammation in this susceptible host, Usp18 +/+ and
Usp18 C61A/C61A - A/J mice were intraperitoneally infected with 1×10⁴ PFU of
CVB3 and sacrificed on day 7 after infection. Echocardiography was conducted at baseline and prior to
sacrifice on day 7 after infection (Fig. 3A).
Enhanced ISGylation was observed in part of the Usp18 C61A/C61A mice on the A/J
background by immunoblotting of protein lysates from cardiac tissues (Fig. 3B). To quantify the degree of
ISGylation, densitometric analysis of the ISG15 smear signal was performed and normalized to tubulin as
loading control. This analysis confirmed a statistically significant increase in protein ISGylation in
Usp18 C61A/C61A mice compared to wild-type controls, consistent with the expected
stabilization of ISGylated proteins due to selective inactivation of the USP18 isopeptidase function. CVB3
infection resulted in significant weight loss and a reduction in body temperature in A/J mice. No
significant differences were detected between Usp18 +/+ and Usp18 C61A/C61A
- A/J animals (Fig. 3C-D). Likewise, blood glucose levels were low at day 7 post-infection, but no
significant differences were observed between genotypes (Fig. 3E). Histological analysis showed pancreatic
destruction in all infected animals, regardless of genotype, supporting successful infection (Fig. 3F).
Furthermore, viral titers and CVB3 RNA levels in the heart did not significantly differ between Usp18
+/+ and Usp18 C61A/C61A - A/J mice (Fig. 3G-H).
All infected animals exhibited cardiac inflammation and immune cell infiltration, as assessed by the
myocarditis score (Fig. 4A). Compared to our results obtained for C57BL/6 mice in this study, myocarditis,
as determined by HE-based scoring of cardiac tissue sections, was not elevated in A/J mice under the
experimental conditions applied here. Consistent with previous reports [46] and despite higher viral load
in infected A/J mice at day 7 post-infection in comparison to C57BL/6 mice at day 8 (Fig. 1E vs. 3H),
histological myocarditis scores averaged around 1.5 in both strains (Fig. 2A vs. 4A). Importantly, like
C57BL/6 mice, no significant differences were detected between Usp18 +/+ and Usp18
C61A/C61A - A/J mice (Fig. 4A). To further characterize immune cell infiltration, flow
cytometry analysis was performed 7 days post-infection in A/J mice (gating strategy provided in
Supplementary Figure S3 and S4). CD45⁺ immune cell infiltration was detected in all infected
animals, with no significant differences between Usp18 +/+ and Usp18
C61A/C61A - A/J mice (Fig. 4B-C). The extent of overall immune cell infiltration in A/J
mice was reduced in comparison to that observed in C57BL/6 mice (Fig. 4B-E vs. 2B-D). This is consistent
with previous studies that also reported that CVB3 infection in A/J mice does not lead to an increase in
immune cell infiltration, despite their higher susceptibility to CVB3 infection, even when assessed at day
8 post-infection [46, 53, 54]. Importantly, shortening the observation period to day 7, as done in this
study, did not appear to impact the detection or overall degree of inflammation in A/J mice, further
supporting the robustness of this model.
We next analyzed the composition of infiltrating immune cells in A/J mice in more detail. Within the
myeloid compartment, no significant differences were found in the frequencies of CD11b⁺Ly6G⁺
neutrophils, CD11b⁺CD11c⁺ dendritic cells, CD11b⁺F4/80⁻ monocytes or
CD11b⁺F4/80⁺ macrophages between Usp18 +/+ and Usp18 C61A/C61A
- A/J animals (Fig. 4D). Within the lymphoid immune cell compartment, no significant differences
were observed in the frequencies of B220⁺ B cells, CD3⁺CD4⁺ T cells or
CD3⁺CD8⁺ T cells between Usp18 +/+ and Usp18 C61A/C61A
animals (Fig. 4E). Within the monocytes, a large percentage of Ly6Chigh inflammatory monocytes
was observed, higher compared to C57BL/6 animals, but no differences were observed between the genotypes
(Fig. 4F-G). Additionally, the expression levels of key inflammatory cytokines and chemokines, Tnf-
α , Il-6 , Il-1 β , Ccl2 , and Cxcl2 , in the heart did not
significantly differ between Usp18 +/+ and Usp18 C61A/C61A - A/J
animals 7 days post-infection (Fig. 4H). Overall, immune cell infiltration and cytokine and chemokine
expression in the heart following CVB3 infection did not show large differences between Usp18
+/+ and Usp18 C61A/C61A - A/J animals, suggesting a minor role of USP18
isopeptidase activity on the inflammatory and immune response in this model system.
To assess the functional impact of CVB3-induced myocarditis, transthoracic echocardiography was performed
at baseline and on day 7 post-infection. This non-invasive imaging modality enables sensitive,
quantitative assessment of cardiac performance in vivo , and is particularly valuable for capturing
early or subtle changes in cardiac function that may not be reflected by immune cell infiltration alone.
The echocardiographic data confirmed the functional impact of CVB3 infection, with clear signs of cardiac
impairment across multiple parameters. While heart rate remained stable and comparable between
measurements (Fig. 5A), end-diastolic and stroke volumes as well as cardiac output declined significantly
following infection (Fig. 5B-D). No significant differences were observed between Usp18
+/+ and Usp18 C61A/C61A - A/J mice (Fig. 5A-D). Ejection fraction showed
a reduction in Usp18 C61A/C61A - A/J mice by day 7, though no significant differences
between genotypes were detected (Fig. 5E). S-wave velocity tended to decrease with infection, but remained
non-significant. Again, no significant differences between wild-type and Usp18 C61A/C61A
- A/J mice were detectable (Fig. 5F). A full overview of echocardiographic measurements is provided in
Table 1. These findings highlight that echocardiography captures the physiological consequences of
inflammation in the beating heart, which may not be apparent through histological or immunological
parameters alone. Importantly, while CVB3 infection clearly compromised cardiac function, increased
ISGylation through USP18 isopeptidase inactivation did not improve cardiac function in this model.
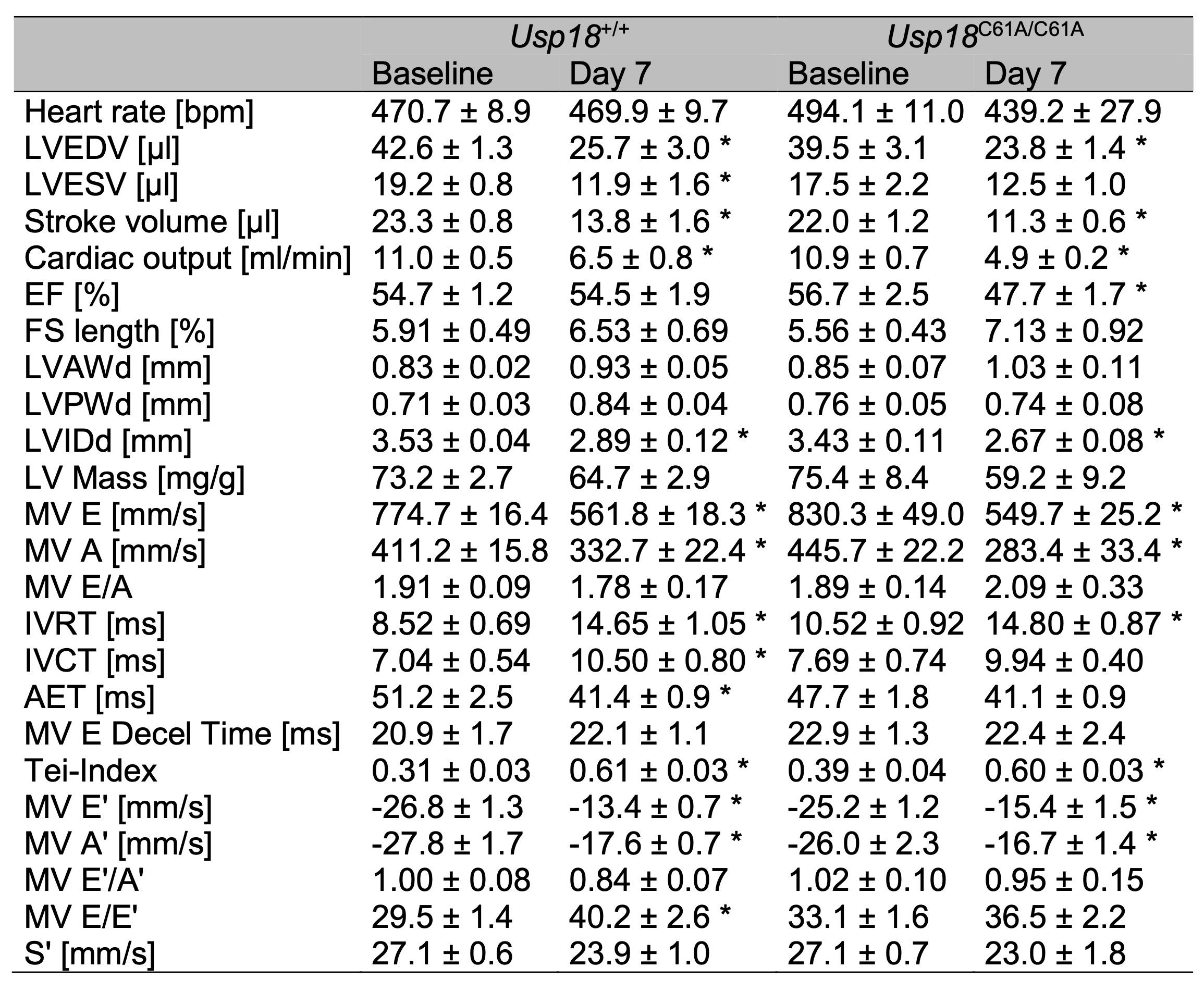
Table 1: Cardiac impairment following CVB3 infection in A/J mice. 7-week-old male Usp18C61A/C61A - A/J mice and Usp18+/+ control mice were intraperitoneally infected with 1 x 104 PFU CVB3. Echocardiography examinations were carried out at baseline (nC61A/C61A = 7, n+/+ = 11) and on day 7 (nC61A/C61A = 5, n+/+ = 11) post infection to evaluate cardiac function. The number of Usp18C61A/C61A - A/J mice analyzed by echocardiography on day 7 post infection was reduced due to poor echocardiographic image quality. Echocardiographic parameters are presented as mean ± SEM. Statistical analysis was conducted using a 2-way ANOVA followed by Tukey's test. Statistically significant results (p < 0.05) are indicated by * for comparisons between baseline and day 7 measurements within each genotype group. Statistical tests comparing Usp18+/+ and Usp18C61A/C61A mice on day 7 post infection have yielded no significant results
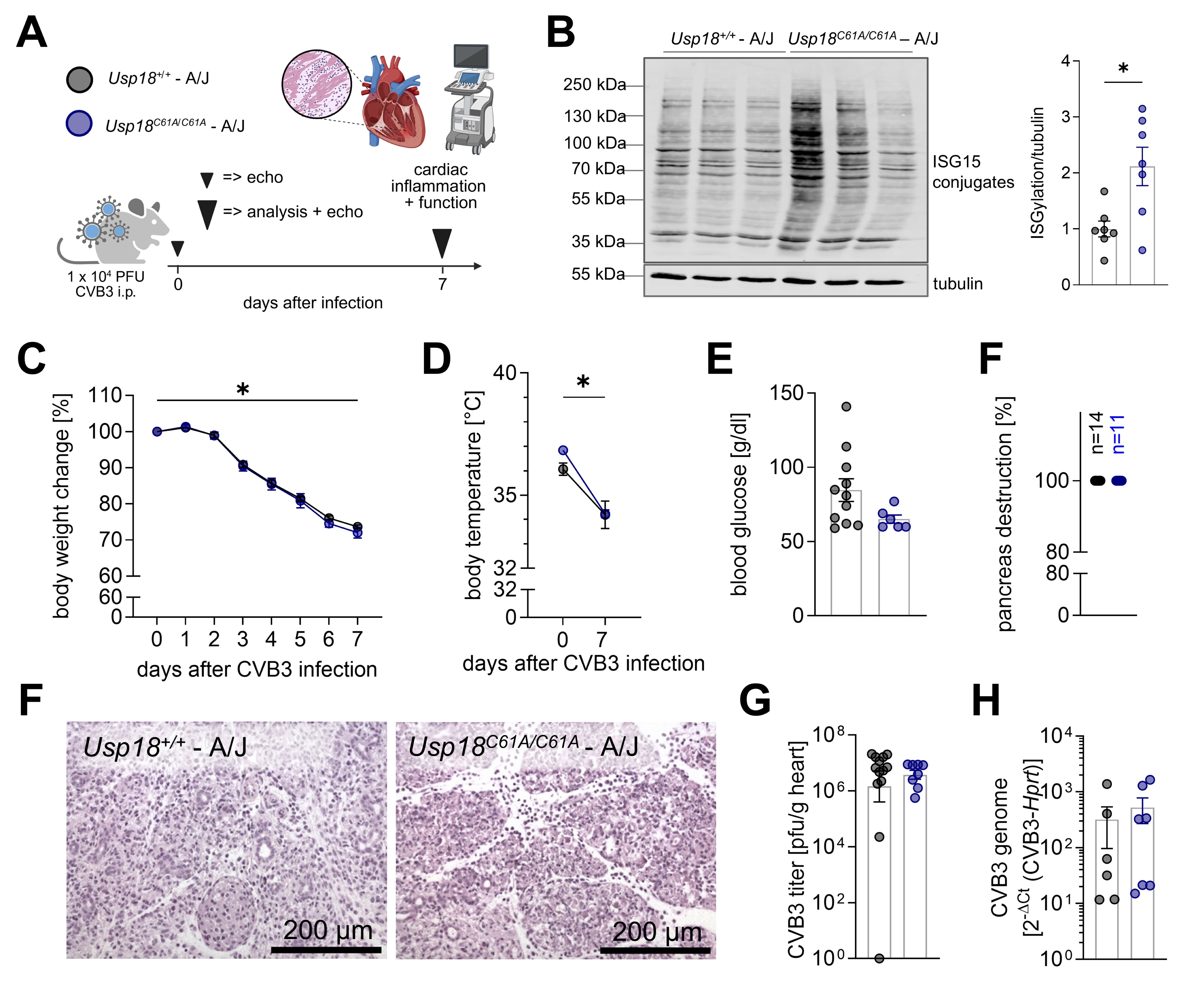
Fig. 3: Systemic conditions and cardiac viral load in Usp18C61A/C61A - A/J mice remain comparable to wild-type controls during CVB3 infection. A: Schematic illustration of the experimental set-up: 7-week-old male Usp18C61A/C61A - A/J mice (nC61A/C61A = 12) and Usp18+/+ control mice (n+/+ = 15) were intraperitoneally infected with 1x 104 PFU CVB3. Animals were analyzed on day 7 post infection to study the effect of enhanced ISGylation on cardiac inflammation. Echocardiographic measurements were carried out at baseline and on day 7 post-infection. B: Detection of ISG15 by Western blot analysis was performed on cardiac tissue homogenates from CVB3 infected mice (nC61A/C61A = 7, n+/+ = 7, n=3 are shown from each genotype) sacrificed on day 7 post infection. Each lane originates from a single animal. Three representative lanes are shown for each genotype. The right panel shows densitometric quantification of ISG15 signal normalized to tubulin. Individual values are presented and expressed as a ratio relative to the mean in Usp18+/+ control mice. Bars represent mean ± SEM. Statistical significance was determined by unpaired t-test; p < 0.05. C: Body weight was measured daily during infection and was normalised to baseline body weight. D: Rectal body temperature was measured during echocardiography at baseline and on day 7 post-infection. Data shown in 4C-D were analyzed using 2-way repeated measures ANOVA (mixed effects model). All time points were compared to baseline regardless of genotype using Dunnett post-hoc test. For clarity, only the results for the comparison baseline – day 7 are depicted in the graphs. Differences between genotypes were tested using Sidak post-hoc tests. E: Serum blood glucose was measured on day 7 post-infection (nC61A/C61A = 6, n+/+ = 11). An unpaired Welch´s t-test revealed no significant results. F: The degree of pancreatic destruction in haematoxylin and eosin (H&E) stained sections was graded by a blinded pathologist (nC61A/C61A = 11, n+/+ = 14). Representative images are shown for both genotypes. The corresponding scale bar (200 µm) is indicated. G-H: Viral load in cardiac tissue was determined by (G) plaque assay (nC61A/C61A = 8, n+/+ = 13) and (H) TaqMan qPCR (nC61A/C61A = 7; n+/+ = 6). The plaque assay data were logarithmically transformed before plotting. Statistical testing was performed using an unpaired Welch´s t-test and an unpaired t-test, respectively. Significant results (p < 0.05) are marked with an asterisk (*), non-significant results are not indicated as such.
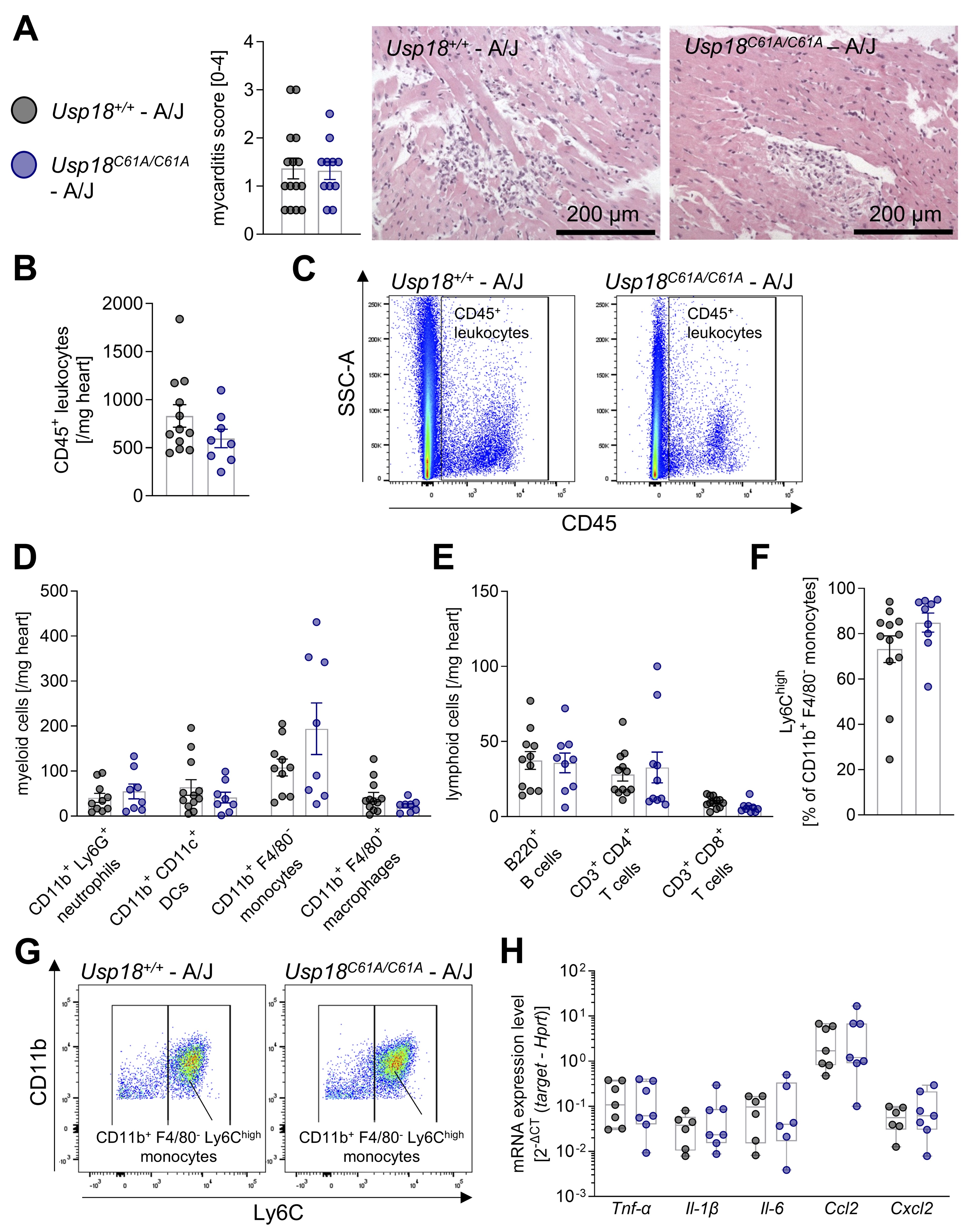
Fig. 4: Enhanced ISGylation is accompanied by ongoing cardiac inflammation in CVB3-infected A/J mice. 7-week-old male Usp18C61A/C61A - A/J mice (nC61A/C61A = 12) and Usp18+/+-A/J control mice (n+/+ = 15) were intraperitoneally infected with 1x 104 PFU CVB3. Animals were analyzed on day 7 post infection to study the effect of enhanced ISGylation on cardiac inflammation. A: Heart tissue samples have been stained with haematoxylin and eosin (H&E) and the myocarditis score was determined (nC61A/C61A = 11, n+/+ = 15). An unpaired t-test showed no statistically significant differences. Representative images are shown for both genotypes. The corresponding scale bar (200 µm) is indicated. B: The CD45+ leukocyte count per milligram cardiac tissue was measured by flow cytometry (nC61A/C61A = 8, n+/+ = 12). C: Representative gating plots of CD45+ leukocytes are depicted for both genotypes. D: The cell count per milligram cardiac tissue was determined by flow cytometry for the following myeloid subpopulations: CD11b+ Ly6G+ neutrophils, CD11b+ CD11c+ dendritic cells (DCs), CD11b+ F4/80- monocytes and CD11b+ F4/80+ macrophages (nC61A/C61A = 8, n+/+ = 12). E: The cell count per milligram cardiac tissue was determined by flow cytometry for the following lymphoid subpopulations: B220+ B cells, CD3+ CD4+ T cells and CD3+ CD8+ T cells (nC61A/C61A = 10, n+/+ = 12) F: The percentage of CD11b+ F4/80- monocytes highly expressing Ly6C (Ly6Chigh) is shown (nC61A/C61A = 9, n+/+ = 12). Data shown in graph 4B – 4F were statistically analyzed by unpaired t-tests, in which each immune cell population was tested separately without correction for multiple comparison. G: Representative gating plots of CD11b+ F4/80- Ly6Chigh monocytes are depicted for both genotypes. H: Cardiac gene expression of chemokines and cytokines have been analyzed with TaqMan qPCR on day 7 post infection (nC61A/C61A = 7, n+/+ = 7). The following target genes have been measured: tumor necrosis factor α (Tnf-α), interleukin-1b (Il-1), interleukin-6 (Il-6) and the chemokines Ccl2 and Cxcl2. Hypoxanthine-guanine phosphoribosyltransferase (Hprt) was used as a housekeeping gene. Results are presented as 2ΔCt (target - Hprt). An unpaired t-test has been performed for each target gene. Significant results (p < 0.05) are marked with an asterisk (*), non-significant results are not indicated as such.
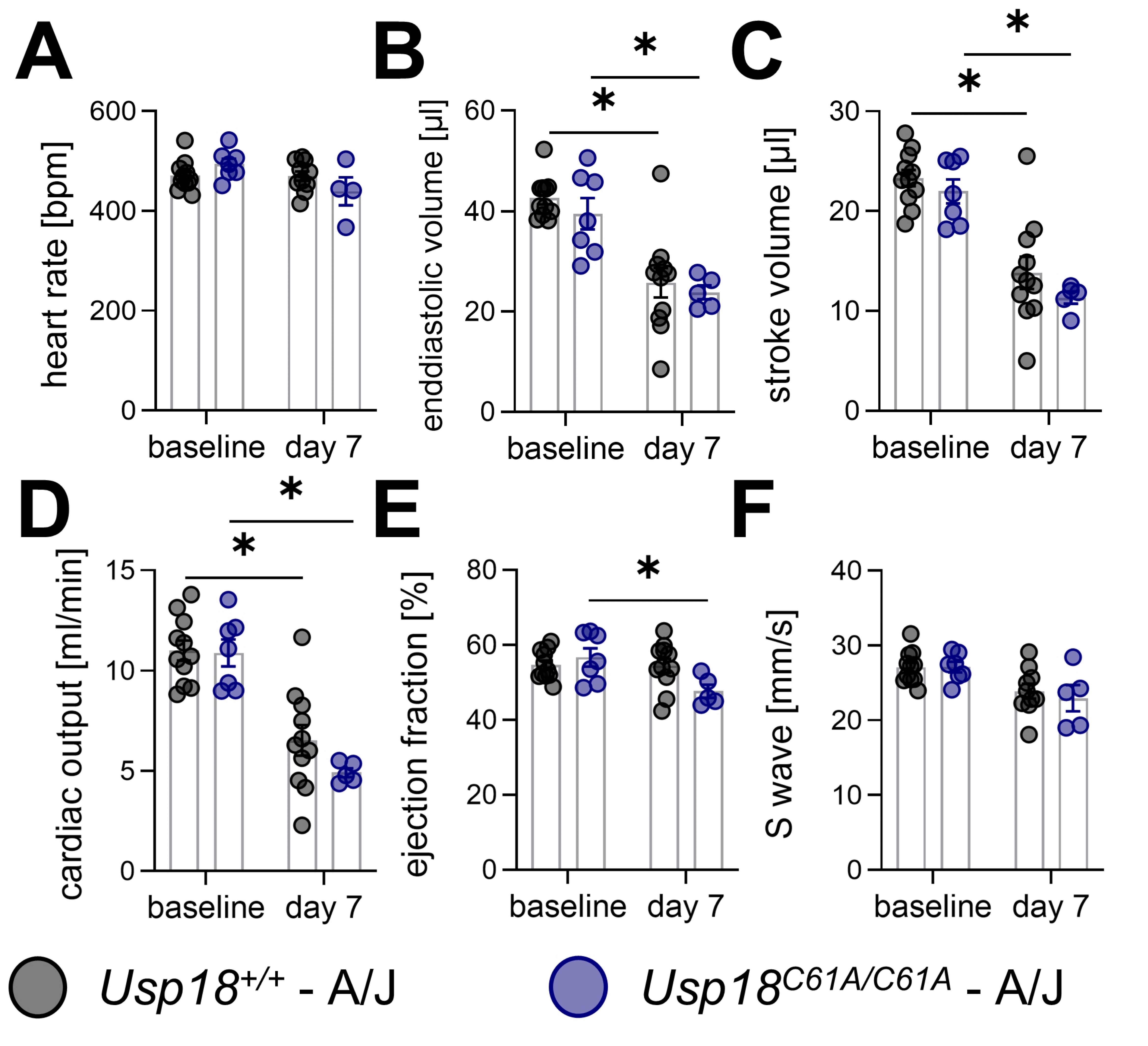
Fig. 5: Cardiac impairment following CVB3 infection remains unchanged in Usp18C61A/C61A - A/J mice. 7-week-old male Usp18C61A/C61A - A/J mice and Usp18+/+ control mice were intraperitoneally infected with 1 x 104 PFU CVB3 and echocardiography examinations were carried out at baseline (nC61A/C61A = 7, n+/+ = 11) and on day 7 (nC61A/C61A = 5, n+/+ = 11) post infection to evaluate cardiac function. The number of Usp18C61A/C61A - A/J mice analyzed by echocardiography on day 7 post infection was reduced due to poor echocardiographic image quality. The following echocardiographic key parameters are presented in graphs: (A) heart rate, (B) enddiastolic volume, (C) stroke volume, (D) cardiac output, (E) ejection and (F) S wave velocity . Data shown in 5A-F were analyzed using 2-way ANOVA. Effects of genotype and time after infection were analyzed using post-hoc Tukey test. Significant results (p < 0.05) are marked with asterisks (*), non-significant results are not indicated as such.
Discussion
The present study investigated whether selective inactivation of USP18’s isopeptidase activity - resulting in enhanced ISGylation - modulates the inflammatory or functional course of CVB3-induced myocarditis. While previous studies have established ISGylation as an essential antiviral effector downstream of type I interferon (IFN) signaling, particularly in non-hematopoietic cells such as cardiomyocytes [9, 32], our data show that stabilized ISGylation alone does not confer measurable benefit during the inflammatory peak of CVB3 myocarditis. In both the moderately affected C57BL/6 background and the more susceptible A/J background, genetic inactivation of USP18’s enzymatic function did not alter immune cell infiltration following infection. The key findings of this study are summarized in the graphical abstract (Fig. 6).
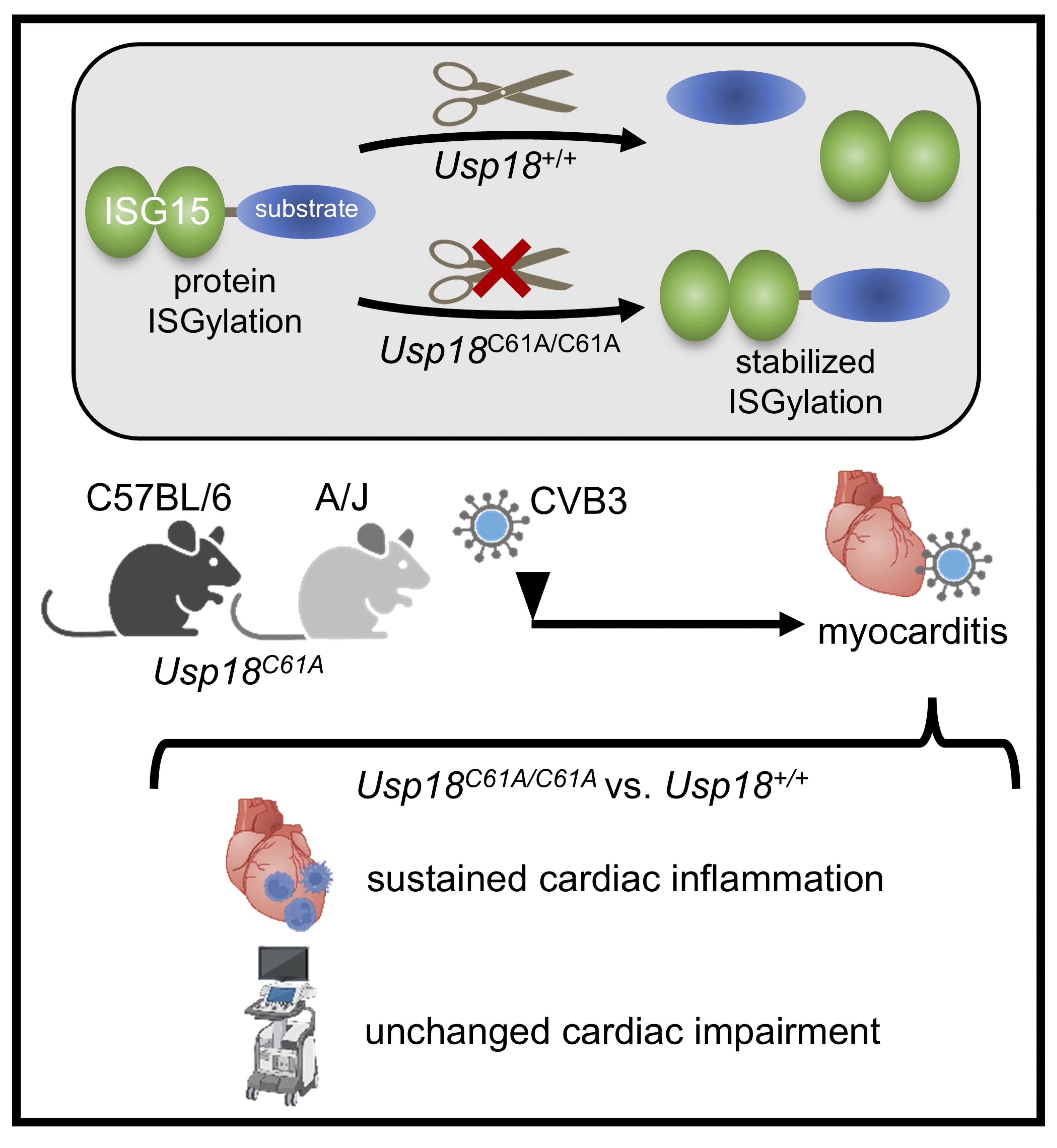
Fig. 6: Graphical abstract. The experimental design and results of the present study investigating enhanced ISGylation in murine CVB3-induced myocarditis are presented: The cysteine-to-alanine substitution in the catalytic core of Usp18 (Usp18C61A/C61A) abolishes its isopeptidase activity, which is crucial for cleaving ISG15 from substrates, thereby stabilising ISGylation. We used Usp18C61A/C61A transgenic mice to study the effects of stabilised ISGylation in the context of Coxsackievirus B3 (CVB3)-induced myocarditis. In both C57BL/6 and A/J background, cardiac inflammation persisted in Usp18C61A/C61A mice compared to Usp18+/+ controls despite increased ISGylation. Furthermore, cardiac impairment assessed by echocardiographic measurements remained unchanged in Usp18C61A/C61A mice compared to wild-type controls.
Previous studies have demonstrated the critical physiological relevance of ISGylation for antiviral defense in the early stages of CVB3 infection. Mice deficient in ISG15 or its E1-activating enzyme Ube1L exhibit higher viral titers and worsened myocardial injury, underscoring the necessity of an intact ISGylation system. Moreover, Usp18 C61A/C61A C57BL/6-mice infected using the same protocol demonstrated increased viral resistance and reduced cardiac viral load at day 6 post-infection, particularly at the level of cardiomyocytes [9]. These findings show that ISGylation plays a central role in establishing early antiviral resistance. However, it remained unclear whether further enhancing this pathway-beyond its endogenous activity-could amplify its protective effects. Our current data show that such enhancement does not translate into improved outcomes during the peak inflammatory phase of CVB3 myocarditis. Instead, our findings support a more nuanced view: although viral cytotoxicity contributes significantly to the inflammatory burden observed around day 7–8 post-infection, and although ISGylation—by limiting viral replication—has a protective effect, further stabilization of this pathway through inactivation of USP18 isopeptidase activity does not confer additional benefit during this peak inflammatory phase. Thus, while ISGylation-mediated viral clearance mitigates early tissue damage and likely influences the extent of subsequent inflammation, immune-mediated injury becomes the predominant pathological driver at later stages. This interpretation is supported by multiple lines of evidence from our data as well as previous studies [55, 56], and reflects a characteristic transition from viral cytotoxicity to immunopathology in enteroviral myocarditis. This decoupling of viral presence and tissue injury highlights the transition from viral damage to immunopathology - a key feature of enteroviral myocarditis models.
Importantly, by using Usp18 C61A/C61A knock-in mice in this study, we selectively analyzed the disabled enzymatic function of USP18 while preserving its ability to negatively regulate IFNAR signaling [37]. The Usp18 C61A/C61A mutation selectively abolishes the enzyme’s isopeptidase activity while leaving its negative regulatory function on type I interferon signaling via IFNAR2–STAT2 interaction intact. In this way, the model enabled specific assessment of the consequences of sustained ISGylation without the confounding effects of amplified IFN responses that are observed in Usp18 -/- mice. This design is supported by previous work showing that Usp18 C61A/C61A knock-in mice exhibit elevated ISGylation and increased resistance to certain viral infections (e.g., influenza B, vaccinia virus), but do not suffer from the deleterious inflammatory phenotypes seen in full USP18 knockouts [37]. Our findings now extend this model to CVB3 myocarditis and suggest that therapeutic targeting of USP18’s isopeptidase function alone may not be sufficient to mitigate cardiac inflammation or dysfunction.
One possible explanation lies in the tissue-specific context of ISGylation. While ISG15 has been shown to shape antiviral responses in various cell types, its role in the heart appears particularly reliant on early, cell-intrinsic antiviral programs within cardiomyocytes. ISGylation stabilizes IFIT1/3 and may act synergistically with these proteins to inhibit viral replication and translation, as shown in vitro [9]. While the current study selectively targets USP18’s isopeptidase function, it does not assess combined modulation of other IFN-stimulated pathways that may act synergistically with ISGylation. The isolated analysis may therefore underestimate the complexity of antiviral defense and inflammatory regulation. Nevertheless, during later stages of myocarditis, when tissue damage is increasingly driven by immune-mediated mechanisms rather than viral cytotoxicity [55, 56], stabilization of ISGylation may have limited impact. This is further supported by our data showing unaltered immune cell profiles and inflammatory cytokine expression in Usp18 C61A/C61A mice at this stage. Of note, we observed a moderate increase in cardiac CD11b⁺CD11c⁺ dendritic cells in Usp18 C61A/C61A mice following CVB3 infection. While this subset included Ly6C⁺ monocyte-derived dendritic cells, which typically expand during inflammation [50, 51], the effect was insufficient to affect the overall immune cell composition or inflammation severity. Whether this reflects a subtle ISGylation-dependent modulation of myeloid cell differentiation or tissue infiltration warrants further mechanistic study.
A central strength of this study lies in the dual-model approach, employing both C57BL/6 and A/J mice. The latter allowed us to test the hypothesis under conditions of more pronounced disease severity. Despite the increased systemic pathology and susceptibility to cardiac autoimmunity in A/J mice [46, 55], the absence of USP18’s isopeptidase activity still did not affect myocarditis progression, suggesting that the limited impact of enhanced ISGylation is not restricted to milder disease contexts. Functionally, echocardiographic analysis confirmed that CVB3 infection in A/J mice impaired cardiac output and stroke volume, primarily due to reduced left ventricular filling. However, these impairments were independent of genotype, indicating that inactivation of USP18 isopeptidase activity does not significantly alter cardiac function during the peak disease phase. This lack of functional difference is consistent with histological and flow cytometry data, further supporting the conclusion that enhanced ISGylation via USP18 inactivation does not provide additional protection in this model of CVB3-induced myocarditis. Even though Usp18 C61A/C61A mice exhibited improved viral control mechanisms earlier in infection [9], these were not sufficient to maintain cardiac function once inflammatory responses were established. On the other hand, our analysis was focused on the acute inflammatory phase of myocarditis (day 7–8 post-infection), and longer-term consequences such as myocardial fibrosis, ventricular remodeling, or chronic dysfunction were not assessed. Although less likely, these later processes may still be influenced by ISGylation, particularly through effects on inflammation resolution or tissue repair. Extended follow-up using longitudinal echocardiographic or histological analyses could help uncover delayed genotype-dependent effects that remain undetectable during the peak of acute inflammation.
Taken together, while ISGylation has been shown to be indispensable for early antiviral defense in CVB3 infection, our data demonstrate that sustained ISGylation via USP18 inactivation does not further attenuate myocardial inflammation or dysfunction during CVB3-induced myocarditis. These findings underscore the complexity of IFN-regulated antiviral programs and suggest that targeting USP18’s enzymatic activity with novel inhibitors may be most effective when timed to early infection phases or combined with additional anti-inflammatory strategies. Further research is needed to define context-specific roles of ISGylation in viral heart disease and to evaluate whether USP18 inhibition could be leveraged therapeutically in other viral or inflammatory conditions.
Resource availability
Materials availability. This study did not generate new unique reagents.
Data and code availability. Any additional information required to reanalyze the data reported in
this paper is available from the lead contact upon request.
Abbreviations
LVEDV (Left Ventricular End-Diastolic Volume); LVESV (Left Ventricular End-Systolic Volume); LVEF (Left Ventricular Ejection Fraction); LVFS (Fractional Shortening of the Left Ventricle); LVAWd (Left Ventricular Anterior Wall in Diastole); LVPWd (Left Ventricular Posterior Wall in Diastole); LVIDd (Left Ventricular Internal Diameter in Diastole); LV (Mass, Left Ventricular Mass); MV (E: Mitral Valve Early Diastolic Wave); MV (A, Mitral Valve Atrial Diastolic Wave); IVRT (Isovolumetric Relaxation Time); IVCT (Isovolumetric Contraction Time); AET ( Aortic Ejection Time); MV (E Decel Time, Mitral Valve E Deceleration Time); MV (E’, Mitral Anulus Early Diastolic Wave); MV (A’, Mitral Anulus Atrial Diastolic Wave); MV (S’, Mitral Anulus Systolic Wave);
Acknowledgements
We acknowledge Anika Lindner, Karolin Voss and Prisca Kunert for excellent technical assistance. We would like to thank the flow cytometry core facility of the German Rheumatism Research Center, Berlin, Germany and of the BIH, Berlin, Germany, for providing access and technical support. AB received support from the Foundation for Experimental Biomedicine Zurich, Switzerland (2016-2023). This project is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 259373024 – TRR 167 (TP B15), Project-ID 318346496 - CRC1292/2 (TP02), project-ID BE 6335/4-3 and BE 6335/5-1 to A.B., Project-ID 437531118 – SFB 1470 (A08) to A.B. Figures were created with images from BioRender, which are subject to the BioRender CC-BY open-access publishing license. The unique image URL is as follows: https://BioRender.com/fd7zwpv (Created in BioRender. Kelm, N. (2025)).
Author contributions
Conceptualization, A.B., K.P.K.; Methodology, N.K., M.K., S.B., K.P.K, K.K.; Investigations, N.K., M.K.,
S.B., S.O. K.K.; Data Analysis, N.K., M.K., S.B., S.O., L.H.; Writing – Original Draft, A.B., N.K.,
L.H.; Writing – Review & Editing, A.B., N.K., L.H.; All co-authors have reviewed the manuscript;
Visualization, N.K., K.K.; Project Administration, A.B.; Funding Acquisition, A.B. N.K. and M.K. equally
contributed to this work.
Statement of Ethics
Animal experiments conform to internationally accepted standards and have been approved by the appropriate
institutional review body.
Disclosure Statement
The authors declare no competing interests.
Declaration of generative AI and AI-assisted technologies
This manuscript was prepared with the assistance of ChatGPT, a language model developed by OpenAI, which was
used for refining language. The authors have reviewed, revised and approved the final content.
References
| 1 | Dalldorf G, Sickles GM: An Unidentified, Filtrable Agent Isolated From the Feces of Children With
Paralysis. Science 1948;108:61-62.
https://doi.org/10.1126/science.108.2794.61 |
| 2 | Kandolf R, Klingel K, Mertsching H, Canu A, Hohenadl C, Zell R, Reimann BY, Heim A, McManus BM,
Foulis AK, et al.: Molecular studies on enteroviral heart disease: patterns of acute and
persistent infections. Eur Heart J 1991;12 Suppl D:49-55.
https://doi.org/10.1093/eurheartj/12.suppl_D.49 |
| 3 | Mena I, Fischer C, Gebhard JR, Perry CM, Harkins S, Whitton JL: Coxsackievirus infection of the
pancreas: evaluation of receptor expression, pathogenesis, and immunopathology. Virology
2000;271:276-288.
https://doi.org/10.1006/viro.2000.0332 |
| 4 | Kim KS, Hufnagel G, Chapman NM, Tracy S: The group B coxsackieviruses and myocarditis. Rev Med
Virol 2001;11:355-368.
https://doi.org/10.1002/rmv.326 |
| 5 | Fairweather D, Stafford KA, Sung YK: Update on coxsackievirus B3 myocarditis. Curr Opin
Rheumatol 2012;24:401-407.
https://doi.org/10.1097/BOR.0b013e328353372d |
| 6 | D'Ambrosio A, Patti G, Manzoli A, Sinagra G, Di Lenarda A, Silvestri F, Di Sciascio G: The fate
of acute myocarditis between spontaneous improvement and evolution to dilated cardiomyopathy: a
review. Heart 2001;85:499-504.
https://doi.org/10.1136/hrt.85.5.499 |
| 7 | Andreoletti L, Bourlet T, Moukassa D, Rey L, Hot D, Li Y, Lambert V, Gosselin B, Mosnier JF,
Stankowiak C, Wattre P: Enteroviruses can persist with or without active viral replication in
cardiac tissue of patients with end-stage ischemic or dilated cardiomyopathy. J Infect Dis
2000;182:1222-1227.
https://doi.org/10.1086/315818 |
| 8 | Leipner C, Grun K, Schneider I, Gluck B, Sigusch HH, Stelzner A: Coxsackievirus B3-induced
myocarditis: differences in the immune response of C57BL/6 and Balb/c mice. Med Microbiol Immunol
2004;193:141-147.
https://doi.org/10.1007/s00430-003-0199-5 |
| 9 | Kespohl M, Bredow C, Klingel K, Voß M, Paeschke A, Zickler M, Poller W, Kaya Z, Eckstein J,
Fechner H, Spranger J, Fähling M, Wirth EK, Radoshevich L, Thery F, Impens F, Berndt N, Knobeloch
KP, Beling A: Protein modification with ISG15 blocks coxsackievirus pathology by antiviral and
metabolic reprogramming. Sci Adv 2020;6:eaay1109.
https://doi.org/10.1126/sciadv.aay1109 |
| 10 | Etchison D, Milburn SC, Edery I, Sonenberg N, Hershey JW: Inhibition of HeLa cell protein
synthesis following poliovirus infection correlates with the proteolysis of a 220, 000-dalton
polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J
Biol Chem 1982;257:14806-14810.
https://doi.org/10.1016/S0021-9258(18)33352-0 |
| 11 | Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE, Knowlton KU: Enteroviral
protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy.
Nat Med 1999;5:320-326.
https://doi.org/10.1038/6543 |
| 12 | Xiong D, Yajima T, Lim BK, Stenbit A, Dublin A, Dalton ND, Summers-Torres D, Molkentin JD,
Duplain H, Wessely R, Chen J, Knowlton KU: Inducible cardiac-restricted expression of enteroviral
protease 2A is sufficient to induce dilated cardiomyopathy. Circulation 2007;115:94-102.
https://doi.org/10.1161/CIRCULATIONAHA.106.631093 |
| 13 | Negishi H, Osawa T, Ogami K, Ouyang X, Sakaguchi S, Koshiba R, Yanai H, Seko Y, Shitara H,
Bishop K, Yonekawa H, Tamura T, Kaisho T, Taya C, Taniguchi T, Honda K: A critical link between
Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc
Natl Acad Sci U S A 2008;105:20446-20451.
https://doi.org/10.1073/pnas.0810372105 |
| 14 | Huhn MH, McCartney SA, Lind K, Svedin E, Colonna M, Flodstrom-Tullberg M: Melanoma
differentiation-associated protein-5 (MDA-5) limits early viral replication but is not essential
for the induction of type 1 interferons after Coxsackievirus infection. Virology 2010;401:42-48.
https://doi.org/10.1016/j.virol.2010.02.010 |
| 15 | Francisco E, Suthar M, Gale M, Jr., Rosenfeld AB, Racaniello VR: Cell-type specificity and
functional redundancy of RIG-I-like receptors in innate immune sensing of Coxsackievirus B3 and
encephalomyocarditis virus. Virology 2019;528:7-18.
https://doi.org/10.1016/j.virol.2018.12.003 |
| 16 | Triantafilou K, Orthopoulos G, Vakakis E, Ahmed MA, Golenbock DT, Lepper PM, Triantafilou M:
Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like
receptor (TLR) 8-dependent. Cell Microbiol 2005;7:1117-1126.
https://doi.org/10.1111/j.1462-5822.2005.00537.x |
| 17 | Mukherjee A, Morosky SA, Delorme-Axford E, Dybdahl-Sissoko N, Oberste MS, Wang T, Coyne CB: The
coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and
apoptotic signaling. PLoS Pathog 2011;7:e1001311.
https://doi.org/10.1371/journal.ppat.1001311 |
| 18 | Feng Q, Langereis MA, van Kuppeveld FJ: Induction and suppression of innate antiviral responses
by picornaviruses. Cytokine Growth Factor Rev 2014;25:577-585.
https://doi.org/10.1016/j.cytogfr.2014.07.003 |
| 19 | Deonarain R, Cerullo D, Fuse K, Liu PP, Fish EN: Protective role for interferon-beta in
coxsackievirus B3 infection. Circulation 2004;110:3540-3543.
https://doi.org/10.1161/01.CIR.0000136824.73458.20 |
| 20 | Althof N, Harkins S, Kemball CC, Flynn CT, Alirezaei M, Whitton JL: In vivo ablation of type I
interferon receptor from cardiomyocytes delays coxsackieviral clearance and accelerates myocardial
disease. Journal of virology 2014;88:5087-5099.
https://doi.org/10.1128/JVI.00184-14 |
| 21 | Koestner W, Spanier J, Klause T, Tegtmeyer PK, Becker J, Herder V, Borst K, Todt D, Lienenklaus
S, Gerhauser I, Detje CN, Geffers R, Langereis MA, Vondran FWR, Yuan Q, van Kuppeveld FJM, Ott M,
Staeheli P, Steinmann E, Baumgartner W, et al.: Interferon-beta expression and type I interferon
receptor signaling of hepatocytes prevent hepatic necrosis and virus dissemination in
Coxsackievirus B3-infected mice. PLoS Pathog 2018;14:e1007235.
https://doi.org/10.1371/journal.ppat.1007235 |
| 22 | Lenschow DJ, Giannakopoulos NV, Gunn LJ, Johnston C, O'Guin AK, Schmidt RE, Levine B, Virgin
HWt: Identification of interferon-stimulated gene 15 as an antiviral molecule during Sindbis virus
infection in vivo. Journal of virology 2005;79:13974-13983.
https://doi.org/10.1128/JVI.79.22.13974-13983.2005 |
| 23 | Knight E, Jr., Fahey D, Cordova B, Hillman M, Kutny R, Reich N, Blomstrom D: A 15-kDa
interferon-induced protein is derived by COOH-terminal processing of a 17-kDa precursor. J Biol
Chem 1988;263:4520-4522.
https://doi.org/10.1016/S0021-9258(18)68812-X |
| 24 | Zhao C, Beaudenon SL, Kelley ML, Waddell MB, Yuan W, Schulman BA, Huibregtse JM, Krug RM: The
UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN-alpha/beta-induced
ubiquitin-like protein. Proc Natl Acad Sci U S A 2004;101:7578-7582.
https://doi.org/10.1073/pnas.0402528101 |
| 25 | Wong JJ, Pung YF, Sze NS, Chin KC: HERC5 is an IFN-induced HECT-type E3 protein ligase that
mediates type I IFN-induced ISGylation of protein targets. Proc Natl Acad Sci U S A
2006;103:10735-10740.
https://doi.org/10.1073/pnas.0600397103 |
| 26 | Ketscher L, Basters A, Prinz M, Knobeloch KP: mHERC6 is the essential ISG15 E3 ligase in the
murine system. Biochem Biophys Res Commun 2012;417:135-140.
https://doi.org/10.1016/j.bbrc.2011.11.071 |
| 27 | Chen L, Sun J, Meng L, Heathcote J, Edwards AM, McGilvray ID: ISG15, a ubiquitin-like
interferon-stimulated gene, promotes hepatitis C virus production in vitro: implications for
chronic infection and response to treatment. J Gen Virol 2010;91:382-388.
https://doi.org/10.1099/vir.0.015388-0 |
| 28 | Kim KI, Giannakopoulos NV, Virgin HW, Zhang DE: Interferon-inducible ubiquitin E2, Ubc8, is a
conjugating enzyme for protein ISGylation. Mol Cell Biol 2004;24:9592-9600.
https://doi.org/10.1128/MCB.24.21.9592-9600.2004 |
| 29 | Oudshoorn D, van Boheemen S, Sanchez-Aparicio MT, Rajsbaum R, Garcia-Sastre A, Versteeg GA:
HERC6 is the main E3 ligase for global ISG15 conjugation in mouse cells. PLoS One 2012;7:e29870.
https://doi.org/10.1371/journal.pone.0029870 |
| 30 | Shi HX, Yang K, Liu X, Liu XY, Wei B, Shan YF, Zhu LH, Wang C: Positive regulation of interferon
regulatory factor 3 activation by Herc5 via ISG15 modification. Mol Cell Biol 2010;30:2424-2436.
https://doi.org/10.1128/MCB.01466-09 |
| 31 | Kim MJ, Hwang SY, Imaizumi T, Yoo JY: Negative feedback regulation of RIG-I-mediated antiviral
signaling by interferon-induced ISG15 conjugation. Journal of virology 2008;82:1474-1483.
https://doi.org/10.1128/JVI.01650-07 |
| 32 | Rahnefeld A, Klingel K, Schuermann A, Diny NL, Althof N, Lindner A, Bleienheuft P, Savvatis K,
Respondek D, Opitz E, Ketscher L, Sauter M, Seifert U, Tschope C, Poller W, Knobeloch KP, Voigt A:
Ubiquitin-like protein ISG15 (interferon-stimulated gene of 15 kDa) in host defense against heart
failure in a mouse model of virus-induced cardiomyopathy. Circulation 2014;130:1589-1600.
https://doi.org/10.1161/CIRCULATIONAHA.114.009847 |
| 33 | Bredow C, Thery F, Wirth EK, Ochs S, Kespohl M, Kleinau G, Kelm N, Gimber N, Schmoranzer J, Voss
M, Klingel K, Spranger J, Renko K, Ralser M, Mulleder M, Heuser A, Knobeloch KP, Scheerer P,
Kirwan J, Bruning U, et al.: ISG15 blocks cardiac glycolysis and ensures sufficient mitochondrial
energy production during Coxsackievirus B3 infection. Cardiovasc Res 2024;120:644-657.
https://doi.org/10.1093/cvr/cvae026 |
| 34 | Malakhova OA, Kim KI, Luo JK, Zou W, Kumar KG, Fuchs SY, Shuai K, Zhang DE: UBP43 is a novel
regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J
2006;25:2358-2367.
https://doi.org/10.1038/sj.emboj.7601149 |
| 35 | Arimoto KI, Lochte S, Stoner SA, Burkart C, Zhang Y, Miyauchi S, Wilmes S, Fan JB, Heinisch JJ,
Li Z, Yan M, Pellegrini S, Colland F, Piehler J, Zhang DE: STAT2 is an essential adaptor in
USP18-mediated suppression of type I interferon signaling. Nat Struct Mol Biol 2017;24:279-289.
https://doi.org/10.1038/nsmb.3378 |
| 36 | Ritchie KJ, Hahn CS, Kim KI, Yan M, Rosario D, Li L, de la Torre JC, Zhang DE: Role of ISG15
protease UBP43 (USP18) in innate immunity to viral infection. Nat Med 2004;10:1374-1378.
https://doi.org/10.1038/nm1133 |
| 37 | Ketscher L, Hannss R, Morales DJ, Basters A, Guerra S, Goldmann T, Hausmann A, Prinz M, Naumann
R, Pekosz A, Utermohlen O, Lenschow DJ, Knobeloch KP: Selective inactivation of USP18 isopeptidase
activity in vivo enhances ISG15 conjugation and viral resistance. Proc Natl Acad Sci U S A
2015;112:1577-1582.
https://doi.org/10.1073/pnas.1412881112 |
| 38 | Basters A, Knobeloch KP, Fritz G: How USP18 deals with ISG15-modified proteins: structural basis
for the specificity of the protease. FEBS J 2018;285:1024-1029.
https://doi.org/10.1111/febs.14260 |
| 39 | Arimoto KI, Miyauchi S, Troutman TD, Zhang Y, Liu M, Stoner SA, Davis AG, Fan JB, Huang YJ, Yan
M, Glass CK, Zhang DE: Expansion of interferon inducible gene pool via USP18 inhibition promotes
cancer cell pyroptosis. Nat Commun 2023;14:251.
https://doi.org/10.1038/s41467-022-35348-5 |
| 40 | Miyauchi S, Arimoto KI, Liu M, Zhang Y, Zhang DE: Reprogramming of tumor-associated macrophages
via NEDD4-mediated CSF1R degradation by targeting USP18. Cell Rep 2023;42:113560.
https://doi.org/10.1016/j.celrep.2023.113560 |
| 41 | Fan JB, Miyauchi S, Xu HZ, Liu D, Kim LJY, Burkart C, Cheng H, Arimoto KI, Yan M, Zhou Y,
Gyorffy B, Knobeloch KP, Rich JN, Cang H, Fu XD, Zhang DE: Type I Interferon Regulates a
Coordinated Gene Network to Enhance Cytotoxic T Cell-Mediated Tumor Killing. Cancer Discov
2020;10:382-393.
https://doi.org/10.1158/2159-8290.CD-19-0608 |
| 42 | Klingel K, Hohenadl C, Canu A, Albrecht M, Seemann M, Mall G, Kandolf R: Ongoing
enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative
analysis of virus replication, tissue damage, and inflammation. Proc Natl Acad Sci U S A
1992;89:314-318.
https://doi.org/10.1073/pnas.89.1.314 |
| 43 | Pinkert S, Kespohl M, Kelm N, Kaya Z, Heuser A, Klingel K, Beling A: Exploration of Analgesia
with Tramadol in the Coxsackievirus B3 Myocarditis Mouse Model. Viruses 2021;13
https://doi.org/10.3390/v13071222 |
| 44 | Szalay G, Sauter M, Hald J, Weinzierl A, Kandolf R, Klingel K: Sustained nitric oxide synthesis
contributes to immunopathology in ongoing myocarditis attributable to interleukin-10 disorders. Am
J Pathol 2006;169:2085-2093.
https://doi.org/10.2353/ajpath.2006.060350 |
| 45 | Zacchigna S, Paldino A, Falcao-Pires I, Daskalopoulos EP, Dal Ferro M, Vodret S, Lesizza P,
Cannata A, Miranda-Silva D, Lourenco AP, Pinamonti B, Sinagra G, Weinberger F, Eschenhagen T,
Carrier L, Kehat I, Tocchetti CG, Russo M, Ghigo A, Cimino J, et al.: Towards standardization of
echocardiography for the evaluation of left ventricular function in adult rodents: a position
paper of the ESC Working Group on Myocardial Function. Cardiovasc Res 2021;117:43-59.
https://doi.org/10.1093/cvr/cvaa110 |
| 46 | Althof N, Goetzke CC, Kespohl M, Voss K, Heuser A, Pinkert S, Kaya Z, Klingel K, Beling A: The
immunoproteasome-specific inhibitor ONX 0914 reverses susceptibility to acute viral myocarditis.
EMBO molecular medicine 2018;10:200-218.
https://doi.org/10.15252/emmm.201708089 |
| 47 | Meyer IS, Goetzke CC, Kespohl M, Sauter M, Heuser A, Eckstein V, Vornlocher HP, Anderson DG,
Haas J, Meder B, Katus HA, Klingel K, Beling A, Leuschner F: Silencing the CSF-1 Axis Using
Nanoparticle Encapsulated siRNA Mitigates Viral and Autoimmune Myocarditis. Frontiers in
immunology 2018;9:2303.
https://doi.org/10.3389/fimmu.2018.02303 |
| 48 | Leuschner F, Courties G, Dutta P, Mortensen LJ, Gorbatov R, Sena B, Novobrantseva TI, Borodovsky
A, Fitzgerald K, Koteliansky V, Iwamoto Y, Bohlender M, Meyer S, Lasitschka F, Meder B, Katus HA,
Lin C, Libby P, Swirski FK, Anderson DG, et al.: Silencing of CCR2 in myocarditis. Eur Heart J
2015;36:1478-1488.
https://doi.org/10.1093/eurheartj/ehu225 |
| 49 | Kelm N, Kespohl M, Smagurauskaite G, Vales S, Karuppanan K, Mburu P, Thiele A, Pinkert S, Bukur
T, Mulleder M, Berndt N, Klingel K, Gaida MM, Bhattacharya S, Beling A: Assessing customized
multivalent chemokine-binding peptide treatment in a murine model of coxsackievirus B3
myocarditis. Basic Res Cardiol 2025;120:393-422.
https://doi.org/10.1007/s00395-025-01098-w |
| 50 | Davison AM, King NJ: Accelerated dendritic cell differentiation from migrating Ly6C(lo) bone
marrow monocytes in early dermal West Nile virus infection. J Immunol 2011;186:2382-2396.
https://doi.org/10.4049/jimmunol.1002682 |
| 51 | Liu Z, Wang H, Li Z, Dress RJ, Zhu Y, Zhang S, De Feo D, Kong WT, Cai P, Shin A, Piot C, Yu J,
Gu Y, Zhang M, Gao C, Chen L, Wang H, Vetillard M, Guermonprez P, Kwok I, et al.: Dendritic cell
type 3 arises from Ly6C(+) monocyte-dendritic cell progenitors. Immunity 2023;56:1761-1777 e1766.
https://doi.org/10.1016/j.immuni.2023.07.001 |
| 52 | Chow LH, Gauntt CJ, McManus BM: Differential effects of myocarditic variants of Coxsackievirus
B3 in inbred mice. A pathologic characterization of heart tissue damage. Lab Invest 1991;64:55-64.
|
| 53 | Goetzke CC, Althof N, Neumaier HL, Heuser A, Kaya Z, Kespohl M, Klingel K, Beling A: Mitigated
viral myocarditis in A/J mice by the immunoproteasome inhibitor ONX 0914 depends on inhibition of
systemic inflammatory responses in CoxsackievirusB3 infection. Basic Res Cardiol 2021;116:7.
https://doi.org/10.1007/s00395-021-00848-w |
| 54 | Bockstahler M, Fischer A, Goetzke CC, Neumaier HL, Sauter M, Kespohl M, Muller AM, Meckes C,
Salbach C, Schenk M, Heuser A, Landmesser U, Weiner J, Meder B, Lehmann L, Kratzer A, Klingel K,
Katus HA, Kaya Z, Beling A: Heart-Specific Immune Responses in an Animal Model of
Autoimmune-Related Myocarditis Mitigated by an Immunoproteasome Inhibitor and Genetic Ablation.
Circulation 2020;141:1885-1902.
https://doi.org/10.1161/CIRCULATIONAHA.119.043171 |
| 55 | Neumann DA, Rose NR, Ansari AA, Herskowitz A: Induction of multiple heart autoantibodies in mice
with coxsackievirus B3- and cardiac myosin-induced autoimmune myocarditis. J Immunol
1994;152:343-350.
https://doi.org/10.4049/jimmunol.152.1.343 |
| 56 | Rose NR: Autoimmunity in coxsackievirus infection. Curr Top Microbiol Immunol 2008;323:293-314.
https://doi.org/10.1007/978-3-540-75546-3_14 |