Original Article - DOI:10.33594/000000839
Accepted 24 November 2025 - Published online
31 December 2025
The Impact of Aging on Organ Systems in Sickle Cell Disease: a Comparative Review of Physiological Adaptation and Dysfunction
Keywords
Abstract
Sickle cell anemia (SCA) is a progressive, systemic disorder that can lead to multi-organ dysfunction. While it has traditionally been most prevalent in regions where malaria is endemic, recent epidemiological studies have shown an increasing disease prevalence in non-endemic areas, primarily attributed to global human migration patterns. The severity of SCA typically worsens with age. In early childhood, affected individuals may present with renal hyperfiltration, neurocognitive delays, cardiac remodeling, and skeletal fragility. The presence of these early manifestations often predicts the development of chronic complications later in life, including splenic atrophy, neurodegeneration, and impaired cerebral perfusion. Adequate management of SCA begins with universal newborn screening programs, enabling early detection and the initiation of appropriate interventions. Therapeutic advancements, ranging from disease-modifying agents such as hydroxyurea to curative options including gene therapy and stem cell transplantation, have significantly improved clinical outcomes; however, long-term morbidity remains a significant challenge. This review aimed to explore the effect of aging on pathophysiological changes and the onset of organ-specific complications in SCA patients. It highlights the importance of age-tailored monitoring and a multidisciplinary approach to detect early signs of organ damage, prevent irreversible complications, and consequently improve overall quality of life.
Introduction
Sickle cell anemia (SCA) is a severe inherited blood disorder transmitted in an autosomal recessive pattern, caused by a single-nucleotide point mutation in the β-globin gene on chromosome 11p15.5, resulting in a monogenic defect that alters the structure and function of hemoglobin [1]. This mutation results in the production of abnormal hemoglobin S (HbS), in which the amino acid glutamic acid (a hydrophilic residue) is replaced by valine (a hydrophobic residue) at the sixth position of the beta-globin chain [2]. Under deoxygenated conditions, HbS undergoes abnormal hydrophobic interactions that promote pathological polymerization of red blood cells (RBCs). This process reduces RBC flexibility, leading to the adoption of the distinctive sickle shape [3, 4].
SCA affects multiple organ systems, with complications often evolving progressively over time. In childhood, early signs of renal, cardiac, neurological, and skeletal involvement may be present. As patients age, the disease burden typically worsens, and complications become more pronounced during adolescence and adulthood. This progressive decline may significantly contribute to morbidity and mortality, driven by splenic atrophy, neurodegeneration, and cardiopulmonary deterioration [5].
This review provides a comprehensive overview of the age-related effects of SCA across various organ systems, emphasizing that organ damage often begins in early childhood and worsens over time. It underscores the importance of implementing continuous and targeted monitoring of affected organs from a young age. Such an approach is crucial for reducing long-term complications and enhancing patient outcomes.
Review Methodology
This narrative review was conducted by searching electronic databases including PubMed, Scopus, ScienceDirect, and Google Scholar. The search covered studies published from January 2000 to August 2025 using combinations of the following keywords: “sickle cell disease,” “sickle cell anemia,” “aging,” “organ dysfunction,” “pediatric,” “adult,” and “long-term complications.”Inclusion criteria were original studies, systematic reviews, and meta-analyses in English that addressed organ-specific effects or age-related pathophysiological changes in SCA. Exclusion criteria included single-case reports, non-English publications, and articles not focusing on age-specific or organ-related outcomes. Reference lists of selected articles were also screened for additional relevant publications to ensure completeness and transparency.
Epidemiology
Sickle cell anemia and its variants are the most common inherited blood disorders, affecting millions of individuals worldwide [6]. While estimates vary, approximately 4.4 million people are affected by SCA globally, and around 300, 000 infants are born with the condition each year, with approximately 3, 000 of those cases occurring in the United States [7]. In the United States, it is estimated that more than 3 million Americans are genetic carriers of SCA and that 80, 000-100, 000 individuals have the disease [8, 9]. According to the Centers for Disease Control and Prevention, SCA predominantly affects individuals of African ancestry, with an incidence of 1 in 365 among Black Americans and 1 in 13, 600 among Hispanic Americans [10]. Although the burden of SCA is global, the disease exhibits marked regional variations. More than 75% of the worldwide burden of SCA occurs in sub-Saharan Africa [11]. These geographical differences in prevalence support the epidemiologic evidence that the sickle cell trait confers a survival advantage against malaria. In malaria-endemic regions, individuals who are heterozygous for the sickle cell gene have a greater likelihood of surviving to reproductive age, perpetuating the gene within affected populations and increasing resistance to malaria. Consequently, SCA is more prevalent in other malaria-endemic regions, including India, the Middle East, the Eastern Mediterranean, and the Arabian Peninsula [12, 13]. However, global human migration has altered disease prevalence; for example, migration from countries with high SCA prevalence has contributed to the introduction and spread of SCA into new populations, such as South Africa, Europe, and the Americas [14].
Despite its widespread geographic distribution, SCA is often underdiagnosed and poorly managed in non-endemic areas, exacerbating disparities in care [15]. Notably, SCA was recognized as a public health priority by the World Health Organization (WHO) in 2006 [16].
Pathophysiology
Sickle RBCs are structurally fragile and exhibit a significantly shortened lifespan, decreasing from 120 days to 10-20 days [17]. Consequently, this reduced lifespan results in chronic anemia due to hemolysis and the release of free hemoglobin, which scavenges nitric oxide (NO), a vital vasodilator and anti-inflammatory mediator. This process promotes vasoconstriction, endothelial dysfunction, oxidative stress, and platelet activation, leading to a hypercoagulable state [18].
In addition to hemolysis, sickled RBCs exhibit abnormal adhesion to the vascular endothelium, resulting in microvascular obstruction and ischemia, which cause vaso-occlusive crises (VOCs), unpredictable and painful episodes primarily affecting the bones, joints, chest, or abdomen, that can be triggered by dehydration, infection, cold exposure, or emotional stress [19]. Recurrent VOCs significantly impair quality of life and are a significant cause of hospital admissions among SCA patients. The chronically heightened inflammatory state of SCA, along with recurrent episodes of vascular damage, contributes to the progression of multi-organ damage [20].
Pediatric to Adult Transition in Sickle Cell Anemia
Renal and Hepatic Complications
SCA often leads to dysfunction of major organs (Table 1, Fig. 1), with the kidneys and liver being the
most
susceptible to impairment due to their high perfusion rates and, consequently, high vulnerability to
microvascular occlusion. Structural changes detected by imaging and biochemical markers of renal
dysfunction can
be observed in pediatric patients with SCA [21].
Evaluation of renal and hepatic biochemical markers revealed significantly lower serum urea and creatinine
levels, along with elevated uric acid levels, in adolescents and young adults (aged 14-26) with SCA
compared to
healthy controls [22], indicating impaired renal and hepatic functions likely attributed to glomerular
hyperfiltration and chronic hemolytic stress [23]. Renal enlargement, particularly of the left kidney, is
a
significant early sign of nephropathy in children with SCA and appears to represent compensatory
hypertrophy due
to glomerular hyperfiltration [21].
In addition to renal and hepatic manifestations, other organ complications include pulmonary infiltrates
and the
development of gallstones, which result from ongoing hemolysis and increased bilirubin turnover [24].

Table 1: Age-Related Clinical Impact of Sickle Cell Anemia on Major Organ Systems
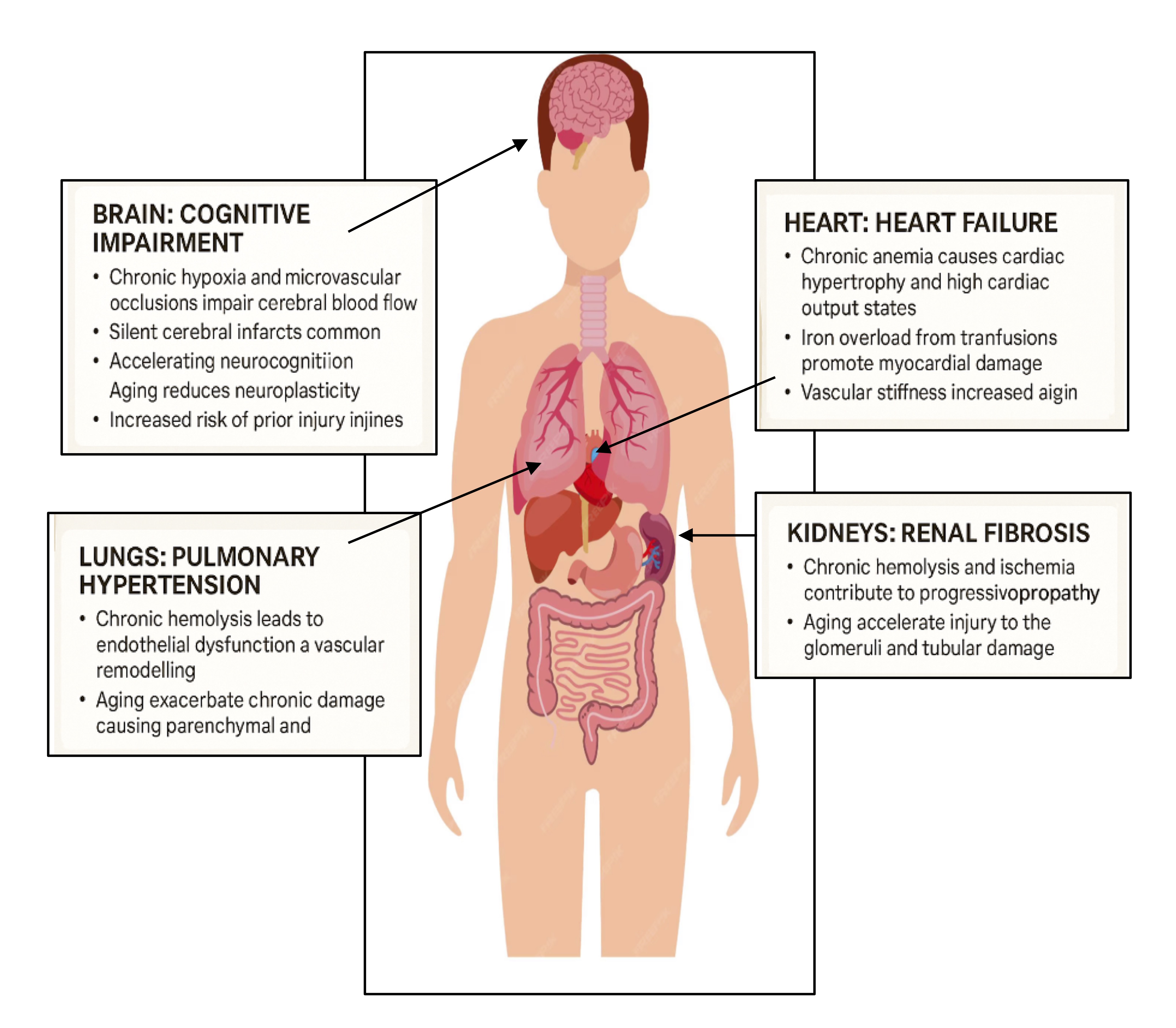
Fig. 1: The impact of aging on organ systems in Sickle Cell Diseases.
Neurological Complications
Neurological complications in SCA manifest early in life and progress throughout the lifespan. Extensive
research has revealed that neurocognitive impairments can occur even in the absence of overt structural
abnormalities on imaging. Despite normal MRI findings, children with SCA exhibit significantly lower IQ
scores,
which may suggest that functional deficits occur prior to visible brain changes [25]. However, a later
decrease
in gray matter volume in pediatric patients was identified, indicating delayed development of structural
changes
[26]. In addition, children with SCA exhibit cognitive performance impairments, manifested by delayed
processing
speed, particularly in males, which is associated with a deterioration of white matter volume. This
finding
supports the predictive role of brain volume changes in determining neurocognitive outcomes in young
children
with SCA [27]. Furthermore, advanced imaging techniques, such as arterial spin labeling, have revealed
alterations in cerebral blood flow and bolus arrival times in pediatric patients (aged 8-18) with SCA,
suggesting age-dependent cerebrovascular adaptations that aim to maintain adequate oxygen delivery to the
brain
[28]. However, these compensatory adaptations appear insufficient to prevent long-term cerebral damage.
With
advancing age, the initial functional and structural deficits observed in pediatric SCA patients often
progress
to more pronounced neurological impairments. In young adults, significant structural degeneration,
including
hippocampal atrophy and reduced intracranial volume, has been reported [29, 30]. Importantly, pediatric
patients
with SCA are at greater risk for developing silent cerebral infarctions or overt stroke [31]. Vasculopathy
of
large vessels is a common underlying cause of stroke in pediatric patients, whereas silent infarctions are
associated with long-term cognitive impairment [32, 33]. Transcranial Doppler ultrasound is widely used as
a
noninvasive screening tool to assess and prevent stroke risk in children [34].
Cardiovascular Complications
Cardiovascular changes in individuals with SCA are evident during childhood. Assessment of cardiac
function in
pediatric patients using M-mode, Doppler, and tissue Doppler echocardiography has revealed cardiomegaly
and
altered myocardial velocities secondary to chronic anemia, which causes volume overload, elevated cardiac
output, and early myocardial stress. This cardiac remodeling is likely a compensatory adaptation to the
altered
hemodynamic demands of SCA in children [35, 36]. However, data on cardiovascular outcomes in adults remain
scarce. This significant gap in the literature should prompt longitudinal studies to determine whether the
early
cardiac changes observed in pediatric patients persist, worsen, or develop into significant clinical
complications later in life.
Spleen Complications
The spleen may develop various complications. It commonly remains functional during early childhood;
however,
repeated episodes of vaso-occlusion and infarction over time often lead to autosplenectomy by adulthood
[37].
Ultrasonographic evaluation of splenic morphology in SCA patients aged 10-52 years showed that 55.4% of
adult
patients had either nonvisible or severely atrophied spleens [38]. The functional asplenia frequently
observed
in adults with SCA significantly increases the risk of severe infections, highlighting the critical need
for
infection prevention strategies [39].
Skeletal Complications
Skeletal complications in SCA often emerge early in life. Patients with SCA below the age of 20 years
demonstrate significantly reduced mandibular bone complexity, indicating early osteopenia or impaired bone
remodeling [40]. Interestingly, mandibular bone abnormalities are less pronounced in older patients,
likely due
to a plateau in bone remodeling or reduced marrow activity over time [40]. The underlying causes of these
skeletal changes may include medullary hyperplasia, bone marrow expansion, and reduced bone mineral
density,
which are common features of chronic hemolytic anemia. Moreover, SCA is associated with bone infarctions,
which
can lead to serious complications such as avascular necrosis, most commonly affecting the head of the
femur
[41].
Diagnosis and Prevention
SCA can be diagnosed at various stages of life, from preconception to adulthood, using a variety of clinical, laboratory, and genetic techniques.
Preembryonic Diagnosis
Preembryonic diagnosis begins even before conception, through the detection of maternal sickle cell
alleles
using polymerase chain reaction (PCR) analysis of the first and second polar bodies of oocytes. This
approach
allows for the selection of mutation-free oocytes prior to fertilization [42].
For couples undergoing in vitro fertilization, preimplantation genetic diagnosis can be performed
on
8-cell embryos from heterozygous parents to identify and select embryos free of the sickle cell mutation
[43].
Ethical and moral concerns regarding the discarding of affected embryos have increased interest in
pre-embryonic
screening techniques to avoid post-implantation decisions [44].
Diagnosis During Pregnancy
During pregnancy, noninvasive prenatal diagnosis is possible through the analysis of cell-free fetal DNA
circulating in maternal plasma. This approach enables the detection of paternally inherited mutations and
mutation dosage. Techniques such as high-resolution melting allow genotyping without the use of labeled
probes,
while more complex genomic regions can be analyzed using unlabeled hybridization probes [45].
Invasive methods, including chorionic villus sampling at 8-12 weeks or amniocentesis at 16 weeks, can be
employed, followed by amplification-refractory mutation system PCR to detect the characteristic β-globin
gene
mutation (GAG→GTG) [46]. However, due to associated risks such as fetal loss, noninvasive methods are
increasingly preferred [47].
At Birth Diagnosis
At birth, newborn screening programs are crucial for early detection before symptoms appear. Selective
screening
is typically limited to newborns whose parents originate from areas with a high prevalence of SCA [48,
49].
Universal screening techniques, including high-performance liquid chromatography, isoelectric focusing,
and
confirmatory DNA analysis, are preferred for the detection of SCA due to their cost-effectiveness and wide
accessibility [50].
SCA should be suspected in patients presenting with characteristic features such as chronic hemolytic
anemia and
frequent VOCs [51]. The diagnosis can be supported by laboratory investigations. Most patients with SCA
exhibit
chronic anemia, with baseline hemoglobin levels often ranging from 6 to 9 g/dL [52]. Laboratory findings
demonstrated elevated reticulocyte counts, high lactate dehydrogenase levels, increased unconjugated
bilirubin,
and low haptoglobin levels, all of which are hallmarks of ongoing hemolysis [53]. Hemoglobin
electrophoresis is
the standard confirmatory test and can distinguish homozygous SCA from other hemoglobinopathies, such as
HbSC or
HbS-β+ thalassemia [50]. Molecular genetic testing provides further precision when needed [50].
Treatment
Although a single-point mutation causes SCA, the disease often manifests as a complex, multisystem disorder that requires a comprehensive, multidisciplinary approach. Effective management often involves hematologists, nephrologists, cardiologists, pain specialists, and mental health professionals [54].
Patients with SCA in high-income countries have experienced improved life expectancy due to advancements in early detection, prophylactic care, and emerging therapies. However, in low-income countries, mortality in early childhood remains high due to limited access to healthcare services [55, 56]. Despite improvements in survival, many patients continue to experience long-term morbidity from chronic complications, multi-organ dysfunction, and psychological stressors, which collectively diminish quality of life [57]. While existing therapies have extended survival, there is a pressing global need to improve access to curative treatments and ensure healthcare equity [58].
Routine management strategies for SCA include folic acid supplementation, prophylactic penicillin in children, and immunization to reduce the risk of infection [59].
Painful VOCs are managed with hydration [60, 61] and non-opioid analgesics for mild to moderate episodes, while more severe cases require opioid analgesics for effective pain control [59]. Blood transfusions can be life-saving for patients with acute severe anemia and may be administered for stroke prevention and perioperative care [62].
Several disease-modifying therapies effectively reduce the burden of SCA. Hydroxyurea increases fetal hemoglobin levels, decreasing the frequency of VOCs and acute chest syndrome, thereby improving overall survival [63, 64]. L-glutamine reduces oxidative stress and lowers the frequency of VOCs [65], while voxelotor inhibits HbS polymerization and increases hemoglobin levels [66-69].
Efforts to cure SCA are making significant progress. Hematopoietic stem cell transplantation is the only established cure, with the best outcomes observed in pediatric patients who have matched sibling donors [70, 71]. Additionally, gene therapy is a rapidly progressing field that may offer promising curative potential [72]. Techniques such as gene addition, gene editing (e.g., CRISPR/Cas9), and reactivation of fetal hemoglobin are under active investigation in clinical trials [73-75].
Future research should focus on advancing personalized treatment strategies and global health initiatives to transform SCA care and ensure accessibility to life-saving interventions for all patients.
Conclusion
SCA follows a progressive, age-dependent course characterized by multi-organ dysfunction that begins in childhood and intensifies over time. Early signs, such as changes in kidney function, neurocognitive development, heart structure, and bone health, often evolve into severe complications, including splenic atrophy, neurodegeneration, and chronic organ failure. This progression is driven by chronic hemolytic anemia and recurrent vaso-occlusive events, leading to cumulative tissue damage. The evidence underscores the need for early, individualized interventions and continuous multi-organ monitoring. Age-specific surveillance is critical for identifying subclinical changes before irreversible damage occurs. However, the lack of long-term data, particularly in adults and elderly patients, highlights the urgent need for longitudinal studies to improve long-term care and quality of life in SCA.
Disclosure Statement
The author declare no competing financial interests.
References
| 1 | Inusa B, Hsu L, Kohli N, Patel A, Ominu-Evbota K, Anie K, et al., Sickle Cell Disease-Genetics,
Pathophysiology, Clinical Presentation and Treatment. Int J Neonatal Screen, 2019. 5(2): p. 20.DOI:
10.3390/ijns5020020.
https://doi.org/10.3390/ijns5020020 |
| 2 | Lattanzi A, Camarena J, Lahiri P, Segal H, Srifa W, Vakulskas C A, et al., Development of β-globin
gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell
disease. Sci Transl Med, 2021. 13(598).DOI: 10.1126/scitranslmed.abf2444.
https://doi.org/10.1126/scitranslmed.abf2444 |
| 3 | Ansari J, Moufarrej Y E, Pawlinski R, and Gavins F N E, Sickle cell disease: a malady beyond a
hemoglobin defect in cerebrovascular disease. Expert Rev Hematol, 2018. 11(1): p. 45-55.DOI:
10.1080/17474086.2018.1407240.
https://doi.org/10.1080/17474086.2018.1407240 |
| 4 | Steinberg M H and Sebastiani P, Genetic modifiers of sickle cell disease. Am J Hematol, 2012.
87(8): p. 795-803.DOI: 10.1002/ajh.23232.
https://doi.org/10.1002/ajh.23232 |
| 5 | Miller S T, Sleeper L A, Pegelow C H, Enos L E, Wang W C, Weiner S J, et al., Prediction of
Adverse Outcomes in Children with Sickle Cell Disease. N Engl J Med, 2000. 342(2): p. 83-89.DOI:
10.1056/nejm200001133420203.
https://doi.org/10.1056/NEJM200001133420203 |
| 6 | Piel F B, Patil A P, Howes R E, Nyangiri O A, Gething P W, Dewi M, et al., Global epidemiology of
sickle hemoglobin in neonates: a contemporary geostatistical model-based map and population
estimates. Lancet, 2013. 381(9861): p. 142-151.DOI: 10.1016/s0140-6736(12)61229-x.
https://doi.org/10.1016/S0140-6736(12)61229-X |
| 7 | Vos T, Allen C, Arora M, Barber R M, Bhutta Z A, Brown A, et al., Global, regional, and national
incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a
systematic analysis for the Global Burden of Disease Study 2015. Lancet, 2016. 388(10053): p.
1545-1602.
https://doi.org/10.1016/S0140-6736(16)31678-6 |
| 8 | Brousseau D C, A Panepinto J, Nimmer M, and Hoffmann R G, The number of people with sickle‐cell
disease in the United States: national and state estimates. Am J Hematol, 2010. 85(1): p. 77-78.DOI:
10.1002/ajh.21570.
https://doi.org/10.1002/ajh.21570 |
| 9 | Hassell K L, Population Estimates of Sickle Cell Disease in the U.S. Am J Prev Med, 2010. 38(4):
p. S512-S521.DOI: 10.1016/j.amepre.2009.12.022.
https://doi.org/10.1016/j.amepre.2009.12.022 |
| 10 | Pokhrel A, Olayemi A, Ogbonda S, Nair K, and Wang J C, Racial and ethnic differences in sickle
cell disease within the United States: From demographics to outcomes. Eur J Haematol, 2023. 110(5):
p. 554-563.DOI: 10.1111/ejh.13936.
https://doi.org/10.1111/ejh.13936 |
| 11 | McGann P T, Hernandez A G, and Ware R E, Sickle cell anemia in sub-Saharan Africa: advancing the
clinical paradigm through partnerships and research. Blood, 2017. 129(2): p. 155-161.DOI:
10.1182/blood-2016-09-702324.
https://doi.org/10.1182/blood-2016-09-702324 |
| 12 | Luzzatto L, SICKLE CELL ANAEMIA AND MALARIA. Mediterr J Hematol Infect Dis, 2012. 4(1): p.
e2012065.DOI: 10.4084/mjhid.2012.065.
https://doi.org/10.4084/mjhid.2012.065 |
| 13 | Esoh K and Wonkam A, Evolutionary history of sickle-cell mutation: implications for global genetic
medicine. Hum Mol Genet, 2021. 30(R1): p. R119-R128.DOI: 10.1093/hmg/ddab004.
https://doi.org/10.1093/hmg/ddab004 |
| 14 | Piel F B, Tatem A J, Huang Z, Gupta S, Williams T N, and Weatherall D J, Global migration and the
changing distribution of sickle haemoglobin: a quantitative study of temporal trends between 1960
and 2000. Lancet Global Health, 2014. 2(2): p. e80-e89.DOI: 10.1016/s2214-109x(13)70150-5.
https://doi.org/10.1016/S2214-109X(13)70150-5 |
| 15 | Musuka H W, Iradukunda P G, Mano O, Saramba E, Gashema P, Moyo E, et al., Evolving Landscape of
Sickle Cell Anemia Management in Africa: A Critical Review. Trop Med Infect Dis, 2024. 9(12): p.
292.DOI: 10.3390/tropicalmed9120292.
https://doi.org/10.3390/tropicalmed9120292 |
| 16 | Organization W H, Sickle-cell Disease: A Strategy for the WHO African Region WHO African Region.
Malabo, Equitorial Guinea, 2010.
|
| 17 | Wang Q and Zennadi R, The Role of RBC Oxidative Stress in Sickle Cell Disease: From the Molecular
Basis to Pathologic Implications. Antioxidants (Basel), 2021. 10(10): p. 1608.DOI:
10.3390/antiox10101608.
https://doi.org/10.3390/antiox10101608 |
| 18 | Kato G J, Steinberg M H, and Gladwin M T, Intravascular hemolysis and the pathophysiology of
sickle cell disease. J Clin Invest, 2017. 127(3): p. 750-760.DOI: 10.1172/jci89741.
https://doi.org/10.1172/JCI89741 |
| 19 | Conran N and Embury S H, Sickle cell vaso-occlusion: The dialectic between red cells and white
cells. Exp Biol Med (Maywood), 2021. 246(12): p. 1458-1472.DOI: 10.1177/15353702211005392.
https://doi.org/10.1177/15353702211005392 |
| 20 | Zadeh F J, Fateh A, Saffari H, Khodadadi M, Eslami Samarin M, Nikoubakht N, et al., The
vaso-occlusive pain crisis in sickle cell patients: A focus on pathogenesis. Curr Res Transl Med,
2025. 73(3): p. 103512.DOI: 10.1016/j.retram.2025.103512.
https://doi.org/10.1016/j.retram.2025.103512 |
| 21 | Nwosu C S, Nri-Ezedi C A, Okechukwu C, Ulasi T O, Umeh E O, Ebubedike U R, et al., Two-Dimensional
Ultrasound Assessment of Long-Term Intra-Abdominal Organ Changes in Children with Sickle Cell Anemia
during Steady State: A Comparative Study. Niger J Clin Pract, 2023. 26(12): p. 1861-1867.DOI:
10.4103/njcp.njcp_411_23.
https://doi.org/10.4103/njcp.njcp_411_23 |
| 22 | Awor S, Bongomin F, Kaggwa M M, Pebolo F P, Epila J, Malinga G M, et al., Liver and renal
biochemical profiles of people with sickle cell disease in Africa: a systematic review and
meta-analysis of case-control studies. Syst Rev, 2024. 13(1).DOI: 10.1186/s13643-024-02662-6.
https://doi.org/10.1186/s13643-024-02662-6 |
| 23 | Allali S, Taylor M, Brice J, and Montalembert M d, Chronic organ injuries in children with sickle
cell disease. Haematologica, 2021. 106(6): p. 1535-1544.DOI: 10.3324/haematol.2020.271353.
https://doi.org/10.3324/haematol.2020.271353 |
| 24 | Chacko S, Jadhav U, Ghewade B, Wagh P, Prasad R, and Wanjari M B, Sickle Cell Disease (SCD)
Leading to Pulmonary Arterial Hypertension (PAH) and Cholelithiasis (CL). Cureus, 2023.DOI:
10.7759/cureus.37113.
https://doi.org/10.7759/cureus.37113 |
| 25 | Steen R G, Fineberg-Buchner C, Hankins G, Weiss L, Prifitera A, and Mulhern R K, Cognitive
Deficits in Children With Sickle Cell Disease. J Child Neurol, 2005. 20(2): p. 102-107.DOI:
10.1177/08830738050200020301.
https://doi.org/10.1177/08830738050200020301 |
| 26 | Hamdule S, Kölbel M, Stotesbury H, Murdoch R, Clayden J D, Sahota S, et al., Effects of regional
brain volumes on cognition in sickle cell anemia: A developmental perspective. Front Neurol, 2023.
14.DOI: 10.3389/fneur.2023.1101223.
https://doi.org/10.3389/fneur.2023.1101223 |
| 27 | Sahu T, Pande B, Sinha M, Sinha R, and Verma H K, Neurocognitive Changes in Sickle Cell Disease: A
Comprehensive Review. Ann Neurosci, 2022. 29(4): p. 255-268.DOI: 10.1177/09727531221108871.
https://doi.org/10.1177/09727531221108871 |
| 28 | Gevers S, Nederveen A J, Fijnvandraat K, van den Berg S M, van Ooij P, Heijtel D F, et al.,
Arterial spin labeling measurement of cerebral perfusion in children with sickle cell disease. J
Magn Reson Imaging, 2012. 35(4): p. 779-787.DOI: 10.1002/jmri.23505.
https://doi.org/10.1002/jmri.23505 |
| 29 | Hamdule S and Kirkham F J, Brain Volumes and Cognition in Patients with Sickle Cell Anaemia: A
Systematic Review and Meta-Analysis. Children (Basel), 2023. 10(8): p. 1360.DOI:
10.3390/children10081360.
https://doi.org/10.3390/children10081360 |
| 30 | Santini T, Koo M, Farhat N, Campos V P, Alkhateeb S, Vieira M A C, et al., Analysis of hippocampal
subfields in sickle cell disease using ultrahigh field MRI. Neuroimage Clin, 2021. 30: p.
102655.DOI: 10.1016/j.nicl.2021.102655.
https://doi.org/10.1016/j.nicl.2021.102655 |
| 31 | Arkuszewski M, Krejza J, Chen R, Ichord R, Kwiatkowski J L, Bilello M, et al., Sickle cell anemia:
Intracranial stenosis and silent cerebral infarcts in children with low risk of stroke. Adv Med Sci,
2014. 59(1): p. 108-113.DOI: 10.1016/j.advms.2013.09.001.
https://doi.org/10.1016/j.advms.2013.09.001 |
| 32 | Switzer J A, Hess D C, Nichols F T, and Adams R J, Pathophysiology and treatment of stroke in
sickle-cell disease: present and future. Lancet Neurol, 2006. 5(6): p. 501-512.DOI:
10.1016/s1474-4422(06)70469-0.
https://doi.org/10.1016/S1474-4422(06)70469-0 |
| 33 | DeBaun M R, Armstrong F D, McKinstry R C, Ware R E, Vichinsky E, and Kirkham F J, Silent cerebral
infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia.
Blood, 2012. 119(20): p. 4587-4596.DOI: 10.1182/blood-2011-02-272682.
https://doi.org/10.1182/blood-2011-02-272682 |
| 34 | Mazzucco S, Diomedi M, Qureshi A, Sainati L, and Padayachee S T, Transcranial Doppler screening
for stroke risk in children with sickle cell disease: a systematic review. Int J Stroke, 2017.
12(6): p. 580-588.DOI: 10.1177/1747493017706189.
https://doi.org/10.1177/1747493017706189 |
| 35 | Olson M, Hebson C, Ehrlich A, New T, and Sachdeva R, Tissue Doppler Imaging-derived Diastolic
Function Assessment in Children With Sickle Cell Disease and Its Relation With Ferritin. J Pediatr
Hematol Oncol, 2016. 38(1): p. 17-21.DOI: 10.1097/mph.0000000000000430.
https://doi.org/10.1097/MPH.0000000000000430 |
| 36 | Ribera M C V, Ribera R B, Koifman R J, and Koifman S, Echocardiography in sickle cell anaemia
patients under 20 years of age: a descriptive study in the Brazilian Western Amazon. Cardiol Young,
2015. 25(1): p. 63-69.DOI: 10.1017/s104795111300156x.
https://doi.org/10.1017/S104795111300156X |
| 37 | Ladu A I, Aiyenigba A O, Adekile A, and Bates I, The spectrum of splenic complications in patients
with sickle cell disease in Africa: a systematic review. Br J Haematol, 2020. 193(1): p. 26-42.DOI:
10.1111/bjh.17179.
https://doi.org/10.1111/bjh.17179 |
| 38 | Ladu A I, Farate A, Garba F, Jeffrey C, and Bates I, 5613357 ULTRASONOGRAPHIC SPLENIC INDICES AND
AGE-RELATED CLINICAL AND LABORATORY PARAMETERS IN PATIENTS WITH SICKLE CELL DISEASE IN NORTH-EASTERN
NIGERIA. Hemasphere, 2023. 7(S1): p. 24-25.DOI: 10.1097/01.hs9.0000928308.87578.05.
https://doi.org/10.1097/01.HS9.0000928308.87578.05 |
| 39 | Obeagu E I and Obeagu G U, Immunization strategies for individuals with sickle cell anemia: A
narrative review. Medicine (Baltimore), 2024. 103(38): p. e39756.DOI: 10.1097/md.0000000000039756.
https://doi.org/10.1097/MD.0000000000039756 |
| 40 | Demirbaş A K, Ergün S, Güneri P, Aktener B O, and Boyacıoğlu H, Mandibular bone changes in sickle
cell anemia: fractal analysis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod, 2008. 106(1): p.
e41-e48.DOI: 10.1016/j.tripleo.2008.03.007.
https://doi.org/10.1016/j.tripleo.2008.03.007 |
| 41 | Almeida-Matos M, Carrasco J, Lisle L, and Castelar M, Avascular necrosis of the femoral head in
sickle cell disease in pediatric patients suffering from hip dysfunction. Rev Salud Publica
(Bogota), 2016. 18(6): p. 986-995.DOI: 10.15446/rsap.v18n6.50069.
https://doi.org/10.15446/rsap.v18n6.50069 |
| 42 | Kuliev A, Rechitsky S, Verlinsky O, Strom C, and Verlinsky Y, Preembryonic diagnosis for sickle
cell disease. Mol Cell Endocrinol, 2001. 183: p. S19-S22.
https://doi.org/10.1016/S0303-7207(01)00569-X |
| 43 | Combs J C, Dougherty M, Yamasaki M U, DeCherney A H, Devine K M, Hill M J, et al., Preimplantation
genetic testing for sickle cell disease: a cost-effectiveness analysis. F S Rep, 2023. 4(3): p.
300-307.DOI: 10.1016/j.xfre.2023.06.001.
https://doi.org/10.1016/j.xfre.2023.06.001 |
| 44 | Kaye D K, Addressing ethical issues related to prenatal diagnostic procedures. Matern Health
Neonatol Perinatol, 2023. 9(1).DOI: 10.1186/s40748-023-00146-4.
https://doi.org/10.1186/s40748-023-00146-4 |
| 45 | Liao G J W, Gronowski A M, and Zhao Z, Non-invasive prenatal testing using cell-free fetal DNA in
maternal circulation. Clin Chim Acta, 2014. 428: p. 44-50.DOI: 10.1016/j.cca.2013.10.007.
https://doi.org/10.1016/j.cca.2013.10.007 |
| 46 | Tan Y, Jian H, Zhang R, Wang J, Zhou C, Xiao Y, et al., Applying amplification refractory mutation
system technique to detecting cell-free fetal DNA for single-gene disorders purpose. Front Genet,
2023. 14.DOI: 10.3389/fgene.2023.1071406.
https://doi.org/10.3389/fgene.2023.1071406 |
| 47 | Goswami A and Patra K K, Non-Invasive Prenatal Testing Versus Invasive Procedures: A Meta-Analysis
of Diagnostic Performance. Eur J Cardiovasc Med, 2025. 15: p. 88-94.
|
| 48 | Thuret I, Sarles J, Merono F, Suzineau E, Collomb J, Lena-Russo D, et al., Neonatal screening for
sickle cell disease in France: evaluation of the selective process: Table 1. J Clin Pathol, 2010.
63(6): p. 548-551.DOI: 10.1136/jcp.2009.068874.
https://doi.org/10.1136/jcp.2009.068874 |
| 49 | Mañú Pereira M and Corrons J L V, Neonatal haemoglobinopathy screening in Spain. J Clin Pathol,
2009. 62(1): p. 22-25.DOI: 10.1136/jcp.2008.058834.
https://doi.org/10.1136/jcp.2008.058834 |
| 50 | Arishi W A, Alhadrami H A, and Zourob M, Techniques for the Detection of Sickle Cell Disease: A
Review. Micromachines (Basel), 2021. 12(5): p. 519.DOI: 10.3390/mi12050519.
https://doi.org/10.3390/mi12050519 |
| 51 | Casale M, Benemei S, Gallucci C, Graziadei G, and Ferrero G B, The phenotypes of sickle cell
disease: strategies to aid the identification of undiagnosed patients in the Italian landscape. Ital
J Pediatr, 2025. 51(1).DOI: 10.1186/s13052-025-01992-y.
https://doi.org/10.1186/s13052-025-01992-y |
| 52 | Rees D C, Brousse V A M, and Brewin J N, Determinants of severity in sickle cell disease. Blood
Rev, 2022. 56: p. 100983.DOI: 10.1016/j.blre.2022.100983.
https://doi.org/10.1016/j.blre.2022.100983 |
| 53 | Feugray G, Dumesnil C, Grall M, Benhamou Y, Girot H, Fettig J, et al., Lactate dehydrogenase and
hemolysis index to predict vaso-occlusive crisis in sickle cell disease. Sci Rep, 2023. 13(1).DOI:
10.1038/s41598-023-48324-w.
https://doi.org/10.1038/s41598-023-48324-w |
| 54 | Powell R E, Lovett P B, Crawford A, McAna J, Axelrod D, Ward L, et al., A Multidisciplinary
Approach to Impact Acute Care Utilization in Sickle Cell Disease. Am J Med Qual, 2018. 33(2): p.
127-131.DOI: 10.1177/1062860617707262.
https://doi.org/10.1177/1062860617707262 |
| 55 | Esoh K, Wonkam-Tingang E, and Wonkam A, Sickle cell disease in sub-Saharan Africa: transferable
strategies for prevention and care. Lancet Haematol, 2021. 8(10): p. e744-e755.DOI:
10.1016/s2352-3026(21)00191-5.
https://doi.org/10.1016/S2352-3026(21)00191-5 |
| 56 | Odame I, Sickle cell disease in children: an update of the evidence in low- and middle-income
settings. Arch Dis Child, 2023. 108(2): p. 108-114.DOI: 10.1136/archdischild-2021-323633.
https://doi.org/10.1136/archdischild-2021-323633 |
| 57 | Tanabe P, Spratling R, Smith D, Grissom P, and Hulihan M, CE: Understanding the Complications of
Sickle Cell Disease. Am J Nurs, 2019. 119(6): p. 26-35.DOI: 10.1097/01.naj.0000559779.40570.2c.
https://doi.org/10.1097/01.NAJ.0000559779.40570.2c |
| 58 | Piel F B, Rees D C, DeBaun M R, Nnodu O, Ranque B, Thompson A A, et al., Defining global
strategies to improve outcomes in sickle cell disease: a Lancet Haematology Commission. Lancet
Haematol, 2023. 10(8): p. e633-e686.DOI: 10.1016/s2352-3026(23)00096-0.
https://doi.org/10.1016/S2352-3026(23)00096-0 |
| 59 | McGann P T, Nero A C, and Ware R E, Current Management of Sickle Cell Anemia. Cold Spring Harb
Perspect Med, 2013. 3(8): p. a011817-a011817.DOI: 10.1101/cshperspect.a011817.
https://doi.org/10.1101/cshperspect.a011817 |
| 60 | Rees D C, Olujohungbe A D, Parker N E, Stephens A D, Telfer P, and Wright J, Guidelines for the
management of the acute painful crisis in sickle cell disease. Br J Haematol, 2003. 120(5): p.
744-752.DOI: 10.1046/j.1365-2141.2003.04193.x.
https://doi.org/10.1046/j.1365-2141.2003.04193.x |
| 61 | Dampier C, Ely E, Eggleston B, Brodecki D, and O'Neal P, Physical and cognitive‐behavioral
activities used in the home management of sickle pain: A daily diary study in children and
adolescents. Pediatr Blood Cancer, 2004. 43(6): p. 674-678.DOI: 10.1002/pbc.20162.
https://doi.org/10.1002/pbc.20162 |
| 62 | Howard J, Sickle cell disease: when and how to transfuse. Hematology 2014, the American Society of
Hematology Education Program Book, 2016. 2016(1): p. 625-631.DOI: 10.1182/asheducation-2016.1.625.
https://doi.org/10.1182/asheducation-2016.1.625 |
| 63 | Cokic V P, Smith R D, Beleslin-Cokic B B, Njoroge J M, Miller J L, Gladwin M T, et al.,
Hydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl
cyclase. J Clin Invest, 2003. 111(2): p. 231-239.DOI: 10.1172/jci16672.
https://doi.org/10.1172/JCI16672 |
| 64 | King S, N-Hydroxyurea and Acyl Nitroso Compounds as Nitroxyl (HNO) and Nitric Oxide (NO) Donors.
Curr Top Med Chem, 2005. 5(7): p. 665-673.DOI: 10.2174/1568026054679362.
https://doi.org/10.2174/1568026054679362 |
| 65 | Sadaf A and Quinn C T, L-glutamine for sickle cell disease: Knight or pawn? Exp Biol Med
(Maywood), 2020. 245(2): p. 146-154.DOI: 10.1177/1535370219900637.
https://doi.org/10.1177/1535370219900637 |
| 66 | Yenamandra P A and Marjoncu P B D, Voxelotor: A Hemoglobin S Polymerization Inhibitor for the
Treatment of Sickle Cell Disease. J Adv Pract Oncol, 2020. 11(8): p. 873-7.DOI:
10.6004/jadpro.2020.11.8.7.
https://doi.org/10.6004/jadpro.2020.11.8.7 |
| 67 | Vichinsky E, Hoppe C C, Ataga K I, Ware R E, Nduba V, El-Beshlawy A, et al., A Phase 3 Randomized
Trial of Voxelotor in Sickle Cell Disease. N Engl J Med, 2019. 381(6): p. 509-519.DOI:
10.1056/nejmoa1903212.
https://doi.org/10.1056/NEJMoa1903212 |
| 68 | Glaros A K, Razvi R, Shah N, and Zaidi A U, Voxelotor: alteration of sickle cell disease
pathophysiology by a first-in-class polymerization inhibitor. Ther Adv Hematol, 2021. 12.DOI:
10.1177/20406207211001136.
https://doi.org/10.1177/20406207211001136 |
| 69 | Hutchaleelaha A, Patel M, Washington C, Siu V, Allen E, Oksenberg D, et al., Pharmacokinetics and
pharmacodynamics of voxelotor (GBT440) in healthy adults and patients with sickle cell disease. Br J
Clin Pharmacol, 2019. 85(6): p. 1290-1302.DOI: 10.1111/bcp.13896.
https://doi.org/10.1111/bcp.13896 |
| 70 | Alshahrani N Z and Algethami M R, The effectiveness of hematopoietic stem cell transplantation in
treating pediatric sickle cell disease: Systematic review and meta-analysis. Saudi Pharm J, 2024.
32(5): p. 102049.DOI: 10.1016/j.jsps.2024.102049.
https://doi.org/10.1016/j.jsps.2024.102049 |
| 71 | Gluckman E, Cappelli B, Bernaudin F, Labopin M, Volt F, Carreras J, et al., Sickle cell disease:
an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation.
Blood, 2017. 129(11): p. 1548-1556.DOI: 10.1182/blood-2016-10-745711.
https://doi.org/10.1182/blood-2016-10-745711 |
| 72 | Obeagu E I, Adias T C, and Obeagu G U, Advancing life: innovative approaches to enhance survival
in sickle cell anemia patients. Ann Med Surg (Lond), 2024. 86(10): p. 6021-6036.DOI:
10.1097/ms9.0000000000002534.
https://doi.org/10.1097/MS9.0000000000002534 |
| 73 | Frangoul H, Altshuler D, Cappellini M D, Chen Y-S, Domm J, Eustace B K, et al., CRISPR-Cas9 Gene
Editing for Sickle Cell Disease and β-Thalassemia. N Engl J Med, 2021. 384(3): p. 252-260.DOI:
10.1056/nejmoa2031054.
https://doi.org/10.1056/NEJMoa2031054 |
| 74 | Abraham A A and Tisdale J F, Gene therapy for sickle cell disease: moving from the bench to the
bedside. Blood, 2021. 138(11): p. 932-941.DOI: 10.1182/blood.2019003776.
https://doi.org/10.1182/blood.2019003776 |
| 75 | Orkin S H and Bauer D E, Emerging Genetic Therapy for Sickle Cell Disease. Annu Rev Med, 2019.
70(1): p. 257-271.DOI: 10.1146/annurev-med-041817-125507.
https://doi.org/10.1146/annurev-med-041817-125507 |