Original Article - DOI:10.33594/000000846
Accepted 4 November 2026 - Published online
30 January 2026
Evidence For a Fibrogenic Interaction Between the Aryl Hydrocarbon Receptor and the Wnt/β-Catenin Pathways in Human Keratinocytes and Fibroblasts
bIUF – Leibniz Research Institute for Environmental Medicine, Düsseldorf, Germany
Keywords
Abstract
Background/Aims:
Systemic sclerosis (SSc) is a rare autoimmune and fibrotic disease, which often manifests in the skin. The aryl hydrocarbon receptor (AHR) is critical for skin homeostasis; however, little is known about its role in fibrosis and SSc. TGFβ, a known target gene of AHR and a fibrogenic cytokine, is also implicated in SSc. Wnt/β-catenin signaling promotes fibrosis, and both the TGFβ and Wnt/β-catenin pathways can act synergistically. Therefore, we investigated the potential triangular crosstalk between TGFβ, AHR, and Wnt/β-catenin signaling in fibrosis.Methods:
Human dermal fibroblasts (HDF) and HaCaT keratinocytes—both “wild-type” and AHR-deficient—were cultured in mono- and co-cultures. Cells were treated with TGFβ, and the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole (FICZ), an AHR agonist. Collagen type I (COL1A1) and matrix metalloproteinase-1 (MMP1) were quantified by ELISA, and Wnt/β-catenin pathway genes were analyzed using ddPCR. Cell migration was assessed using the scratch assay, and proteome profiling was performed for secreted factors.Results:
AHR deletion in HDF reduced Wnt/β-catenin pathway gene expression, but adding an AHR ligand did not further increase the expression of these genes. AHR deficiency in HDF abrogated TGFβ-induced collagen production in monocultures, as did the presence of HaCaT cells in co-cultures. A proteome profile and KEGG analysis of co-cultures showed AHR-dependent regulation of immune-related genes. Finally, scratch closure was also AHR-dependent in both cell types, and this effect could not be fully rescued by TGFβ addition.Conclusion:
This study highlights a context-dependent role of AHR in skin fibrosis and a complex triangular relationship with TGFβ and Wnt/β-catenin signaling. More research is needed to evaluate AHR as a potential therapeutic target in SSc.Introduction
Fibrosis is a severe pathological process characterized by excessive extracellular matrix (ECM) deposition, most commonly affecting the skin, lungs, and gastrointestinal tract. Systemic sclerosis (SSc) is an autoimmune connective tissue disease characterized by fibrosis of multiple organs, including the skin. SSc is a debilitating disease, and therapeutic options remain limited partly due to the disease’s complexity, but also because of the gaps in knowledge regarding the underlying molecular biology. Several major fibrogenic players are known, in particular TGFβ and Wnt/β-catenin signaling, which can regulate the balance of collagen secretion and degradation. Less considered is the aryl hydrocarbon receptor (AHR) as an inducible transcription factor that possibly links to TGFβ and fibrosis [1, 2]. In the skin, crosstalk between keratinocytes and fibroblasts is important for maintaining skin homeostasis [3–5]. Fibroblasts secrete and provide extracellular matrix (ECM) molecules, i.e. collagens, which in turn are degraded by matrix metalloproteinases (MMPs), balancing and structuring the ECM. In connective tissue diseases, fibroblasts produce excess ECM and adhere more to it, leading to increased tissue stiffness [6]. MMP levels are regulated by endogenous inhibitors known as tissue inhibitors of metalloproteinases (TIMPs) [7], and ECM seems to depend on keratinocyte-derived cytokines, such as IL-1β [3, 5].
A key player in ECM homeostasis is transforming growth factor (TGF)-β, which can aberrantly activate fibroblasts to produce abundant collagen. TGFβ is produced mainly by fibroblasts and immune cells in the skin. Indeed, a simple and recognized in vitro fibrosis model is the addition of TGFβ to fibroblasts, which vastly enhances collagen production [8]. However, it is also recognized that Wnt/β -catenin signaling pathway contributes to pathological fibrosis [9, 10]. As the starting point of the signaling, Wnt binds to the surface receptor frizzled, which stops the constantly ongoing degradation of cytoplasmic β-catenin and allows its translocation to the nucleus. There, β-catenin acts as a transcriptional co-factor for various target genes, including fibrogenic genes such as collagen [11, 12]. Wnt signaling can be inhibited by, e.g., WIF1 or DKK2 upregulation, which prevents β-catenin stabilization in the cytoplasm [13]. Interestingly, TGFβ modulates Wnt/ β-catenin signaling at various points, promoting both Wnt production and enhancing β-catenin activation [14].
The AHR is known to be involved in skin physiology [15, 16]. Interestingly, skin fibroblasts from patients with SSc have been reported to exhibit reduced AHR levels, and the authors thus suggested a potential anti-fibrotic role of the AHR [17]. Murai et al. (2018) demonstrated in normal human dermal fibroblasts that the up-regulation of the ECM degrading MMP1 enzyme may occur in an AHR-dependent manner [7], which was also found in orbital fibroblasts [18]. While increased MMP1 levels have been observed in other autoimmune conditions such as rheumatoid arthritis [19], biopsies from SSc patients have also shown decreased MMP1 [20, 21], showing disease-specific regulation.
Mechanistically, TGFβ can suppress AHR transcription, while the AHR can modulate TGFβ signaling via interactions with key regulatory proteins. The AHR is implicated in the Wnt/β-catenin pathway as well [22–25], because apparently AHR activation antagonizes Wnt/β-catenin signaling [26–28]. While AHR suppresses pro-fibrotic behavior in fibroblasts, and robust anti-Wnt mechanisms are established in other cells, a direct and functionally verified connection between AHR and Wnt/β-catenin signaling in mammalian fibroblasts remains to be demonstrated.
In this study, we therefore investigate a potential triangular crosstalk between the Wnt/β-catenin, AHR, and TGFβ signaling pathways in human dermal fibroblasts and keratinocytes using AHR-deficient cells. Additionally, we examine keratinocyte-fibroblast interactions under fibrotic conditions using in vitro co-culture models.
Materials and Methods
Cell Monoculture
We used HaCaT cells, a spontaneously immortalized, non-tumorigenic human
keratinocyte line [29], and human dermal fibroblasts (HDF). AHR was deleted in HaCaT by CRISPR/Cas9 in the
IUF’s
in-house facility, described previously [30], to generate AHR-KO cells. HDF and an AHR-deficient clone
thereof,
DU96 (referred to as HDF-KO in this manuscript), were cultured at 37oC and 5% CO2 in
DMEM
medium (Pan Biotech, cat. no. P04-03590) supplemented with 10% FBS (Pan Biotech, cat. no.
P40-37500) and antibiotics (penicillin and streptomycin). Cells were seeded in six-well plates at a
density of 2x105 cells per well in 1 ml. When the cells reached 70% confluence, they were
treated
with reagents and incubated for 24h and 48h. Similarly, HaCaT cells were seeded at a density of
5x105
cells per well and cultured until they reached 70% confluence in DMEM medium as described above. After
reaching
confluence, both HDF and HaCaT cells were treated with 100 nM 6-formylindolo[3, 2-b]carbazole
(FICZ,
MedChem Express LLC, USA) or TGFβ (Gibco, cat. no. PHG9214) at a concentration of 10 ng/ml or 25
ng/ml,
and incubated for 24 h and 48 h. Cell pellets were preserved in 500 µl of RNAlater (Invitrogen, cat no.
AM7022) and stored at -80°C for further analysis. Supernatants from the monoculture experiment were
collected for subsequent ELISA tests. The number of replicates is indicated in the figure legends.
Co-cultures setup
HDF and HaCaT cells (and their respective AHR-KO variant) were co-cultured using a transwell insert system
with
a 0.4 µm pore diameter (Merck, cat. no. PTHT06H48). HDF cells were seeded at the bottom of six-well
plates at a density of 2 × 10⁵ cells per well, while HaCaT cells were independently seeded onto hanging
cell
culture inserts at a density of 5 × 10⁵ cells per insert to allow for confluence. Cells were incubated at
standard conditions until reaching approximately 70% confluence. Upon reaching the desired confluence
(after
approx. 24 h), the inserts containing either HaCaT or HaCaT-KO cells were carefully transferred into wells
containing the corresponding HDF populations to establish the co-culture and incubated for 48 h in
standard
conditions. This resulted in two experimental groups, consisting of HDF and HaCaT cells with AHR or their
AHR
impaired variant. All conditions were performed in triplicate. Supernatants from the co-culture experiment
were
collected for subsequent ELISA (MMP1 and COL1A1) and Proteome Profiler analyses.
COL1A1 and MMP1 measurement by ELISA
Commercial ELISA kits were used to quantify collagen type I (RayBiotech, cat. no. ELH-COL1A1) and
MMP1 in
cell culture supernatants (RayBiotech, cat. no. ELH-MMP1). Tests were performed according to the
manufacturer’s instructions. All samples were analyzed in triplicate.
Gene expression
Gene expression analysis was conducted on cultured cells from monoculture experiments only. RNA from ~2
x105 HDF and ~ 1 x106 HaCaT cells was isolated using RNeasy Micro RNA Kit (Qiagen,
cat.
no. 74004). The quantity and quality of isolated RNA were evaluated on a DeNovix spectrophotometer.
RNA
purity was determined based on the 260/280 nm OD ratio, with expected values between 1.8 and 2.0. RNA was
stored
in ultra-clean, RNase-free water provided by the manufacturer at -80°C until further analysis.
Reverse transcription was performed using the High Capacity cDNA Reverse Transcription Kit (Applied
Biosystems,
Carlsbad, CA, USA), according to the manufacturer’s instructions. ddPCR Supermix for Probes (no dUTP;
Bio-Rad,
Hercules, CA) and TaqMan probes (Thermo Fisher Scientific, USA) were used for the gene expression
analysis:
AHR (Hs00169233_m1), FZD2 (Hs00361432_s1), DKK2 (Hs00205294_m1), TCF7
(Hs01556515_m1), LEF1 (TCF10, Hs01547250_m1), WIF1 (Hs00183662_m1), and CTNNB1
(Hs00164383_m1). These genes, except AHR, are hereafter referred to as “Wnt/β-catenin pathway
genes”.
Each reaction was then loaded into the sample well of an eight-well disposable cartridge (Bio-Rad) along
with 70
µl of droplet generation oil (Bio-Rad), and droplets were formed in a final volume of 40 µl using the
Droplet
Generator (Bio-Rad). Droplets were transferred to a 96-well PCR plate, heat-sealed with foil, and
amplified
using a C1000 Touch Thermal Cycler (Bio-Rad). Results expressed in copies/µl were converted into
copies/ng. Gene
expression analysis was performed in three independent experiments, with each sample analyzed in
triplicate. To
determine if the incubation time (24 or 48 hours) influenced the effect of other predictive variables on
gene
expression, regression models incorporating an interaction term between time and these variables were
employed.
Although a subset of samples showed higher gene expression after 48 hours of incubation compared to 24
hours,
the regression model did not reveal any significant interaction between time and the effect of other
variables
(p > 0.05, data not shown). Furthermore, interaction plots visually confirmed the absence of such an
effect.
This result indicates that the incubation time did not significantly modulate the impact of the other
variables
on gene expression. Consequently, results for both incubation times are presented together in the final
gene
expression figures and graphs.
Proteome profiler
For the analysis of proteins secreted by HDF and HaCaT in co-cultures, the Proteome
Profiler Human XL Cytokine Kit (cat. no. ARY022B, R&D Systems, USA) was used. A total of 500 µl
of
supernatant from the WT/WT co-culture and 500 µl from the KO/KO co-culture were analyzed according to the
manufacturer's protocol. Specifically, antibody-labeled membranes provided by the manufacturer were
incubated in
a kit-provided buffer for one hour at room temperature. After removing the buffer, the supernatants -
diluted in
the same buffer - were added and incubated overnight at 2–8°C. The following day, the protocol was
continued as
per the manufacturer's instructions, resulting in membranes with bound proteins. Chemiluminescence
analysis was
performed using the iBright CL750 imaging system (Thermo Fisher Scientific, USA), with the membrane
exposure
time automatically set by the device software to approximately 30 minutes. Signal intensities were
visually
scored using a 4-point scale: 0 = no expression, 1 = weak (+), 2 = moderate (++), and 3 = strong (+++).
Differences in signal intensity between KO/KO and WT/WT groups were calculated and visualized as (KO/KO –
WT/WT)
(Table S2). To validate the findings obtained from the Proteome Profiler, we further quantified IL-8
secretion
using an independent ELISA assay (cat. no. KE00006, Proteintech) according to manufacturer's
instructions.
Proteins showing differential expression were subjected to KEGG pathway enrichment analysis using the
clusterProfiler R package [31] and the org.Hs.eg.db annotation database [32], in order to identify
biological
pathways most affected by AHR deletion. Data visualization, including signal-intensity plot, was performed
using
the ggplot2 package [33] in R. Pathways were categorized based on the number of upregulated (positive
signal
difference) and downregulated (negative signal difference) proteins in KO/KO cells compared to WT/WT.
Scratch assay
Cells were seeded in 24-well culture plates at a high density: 1x105 cells for HaCaT and 8
x104
cells for HDFs and cultured under standard conditions. After overnight incubation, when the cells
reached
95% confluence, the cell layers were scratched diagonally in the middle of each well using a 200 µl
pipette tip.
After scratching, cells were washed with PBS four times to remove the debris formed by scratching. Cells
were
treated with TGFβ 25 ng/ml, FICZ 100 nM, or a combination of TGFβ 25 ng/ml + FICZ 100 nM. In the case of
HDF
cells (Fig. 6C), additional conditions included treatment with the AHR inhibitor CH223191 (10 µM), either
alone
or in combination with TGFβ. Images were taken at 0 h, 24 h and 30 h or 48 h after scratching. To assess
the
effects of AHR impairment in HDF cells, we used the AHR antagonist CH223191 to block AHR signaling, as
HDF-KO
cells were no longer available for these experiments. Serum-free DMEM medium was changed daily with the
appropriate treatment. Light microscope images with a 40x magnification were taken right after scratching,
as
well as after 20 h and 30 h of incubation in case of HDF cell and 24 h and 48 h in the case of HaCaT
cells. The
images were then analyzed using ImageJ wound healing size tool, an automated option which, after manual
corrections, allowed the measurement of the area of the wound gap in percentage of the whole photographed
area.
Statistical analysis of gene expression data
Statistical analyses and data visualizations were performed in the R environment (R Core Team, 2024). The
following R packages were used: tidyverse, readxl, ggpubr, cowplot, flexplot, and GGally. To assess the
effect
of incubation time and its interactions with other variables on gene expression, a generalized regression
model
was applied. Correlation analysis was performed on logarithmically transformed gene expression data using
the
ggpairs function from the GGally R package. The results were presented as a correlation plot matrix,
including
both visual representations of the correlations and numerical values of the correlation coefficients with
their
corresponding p-values. Comparisons of expression levels between groups were conducted using
non-parametric
statistical tests: the Wilcoxon test for pairwise comparisons and the Kruskal-Wallis test for multiple
group
comparisons. Due to the exploratory nature of the study, correction for multiple testing was not applied.
The
aim was to identify potential patterns and hypotheses for future validation rather than to draw definitive
inferential conclusions. A p-value < 0.05 was considered statistically significant.
Statistical analysis of ELISA data
GraphPad Prism software (version 10.1.2; GraphPad Software, San Diego, CA, USA) was used for analysis and
data
visualization. Two-way ANOVA followed by a post hoc multiple comparisons test was used to analyze
differences
between groups. A p-value < 0.05 was considered statistically significant.
Results
Absence of AHR modulates gene expression of Wnt/β-catenin genes – downregulation in fibroblasts,
upregulation
in HaCaT
We performed digital droplet PCR with RNA isolated from AHR-proficient and AHR-deleted HDF and HaCaT cells
to
test whether AHR affects the expression levels of the Wnt/β-catenin pathway genes frizzled2 (FZD2),
Wnt
inhibitory factor (WIF1), dickkopf inhibitor 2 (DKK2), lymphoid enhancer binding factor 1
(LEF1), transcription factor 7 (TCF7), and β-catenin 1 (CTNNB1).
As shown in Fig. 1A and supplementary Figure S1, the absence of AHR in fibroblasts resulted in a
significantly
lower expression of all investigated Wnt/β-catenin pathway genes. WIF1 and LEF1 transcript
levels
were below the limit of detection and are therefore not shown. However, DNA methylation analysis for these
genes
is provided in the supplementary material, suggesting that epigenetic regulation may contribute to their
silencing (Figure S2-S5). Except for DKK2 expression, AHR deficiency in HaCaT cells resulted in
upregulation of these pathway genes, suggesting that AHR suppresses their expression (Fig. 1B). Again,
WIF1 and LEF1 levels were below the limit of detection, irrespective of the presence or
absence of
AHR.
Of note, the overall expression levels of FZD2 and CTNNB1 were similar in both cell types.
Notably, DKK2 expression was higher in both WT cell lines. Together, the data indicate for the
first time
that the presence of AHR is needed for basal expression of selected Wnt/β-catenin pathway genes and
β-catenin
expression itself in HDFs. In contrast, presence of AHR appears to have a dampening effect on the
expression of
these genes in HaCaT cells.
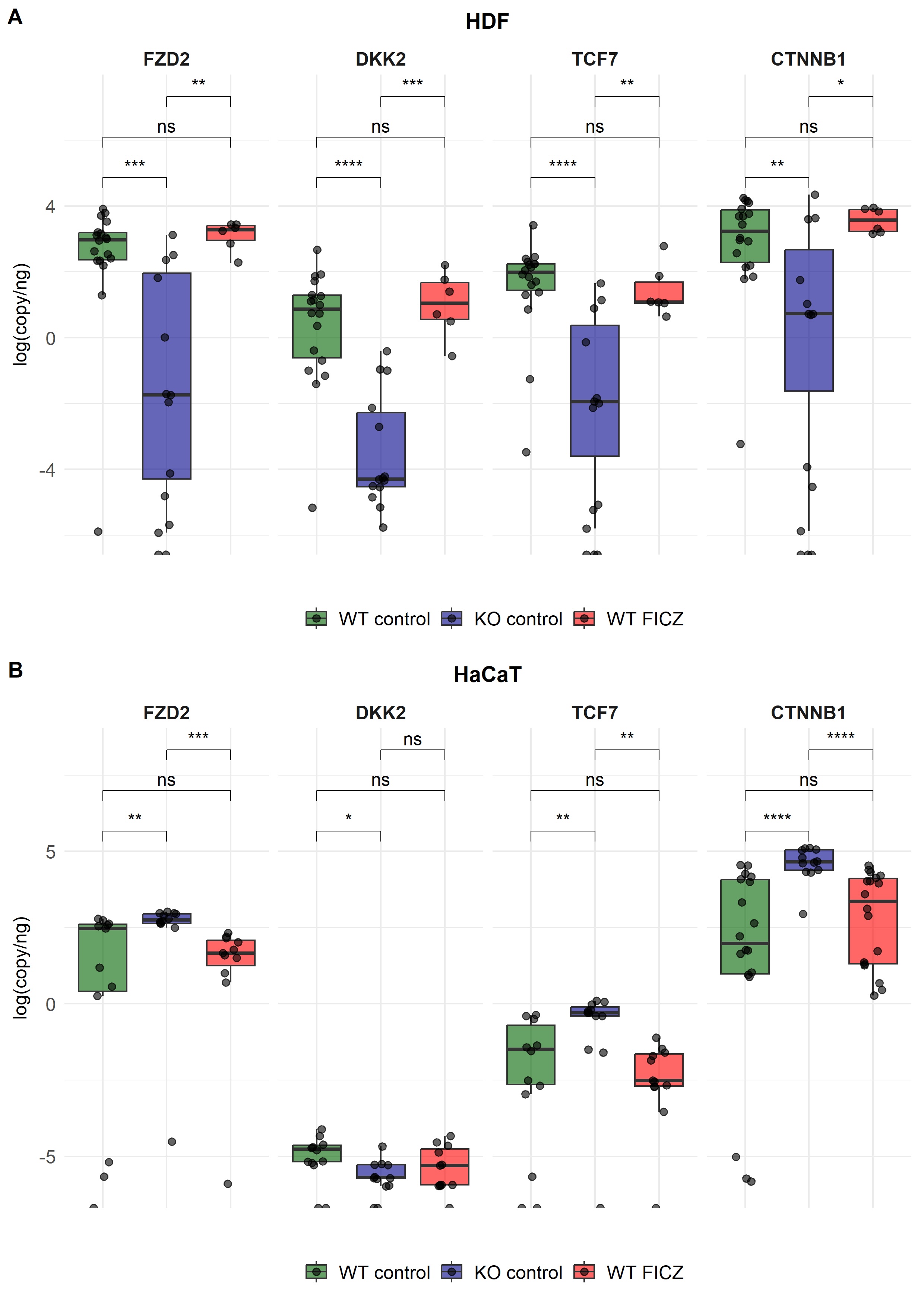
Fig. 1: Expression of selected Wnt/β-catenin pathway genes measured by ddPCR, presented as log-transformed copies/ng in human dermal fibroblasts (HDF, panel A) and HaCaT, (panel B). Green, blue, and red boxplots with individual data points represent “wild-type” (WT), AHR-knockout (KO) cells, and WT cells treated with the AHR agonist FICZ (red), respectively. Cells were collected from three independent experiments, and ddPCR results were pooled from two timepoints (24 h and 48 h). All samples were performed in triplicate. Statistical comparisons were performed using the Wilcoxon test. *: p <= 0.05; **: p <=0.01; ***: p <=0.001; ****: p <=0.0001; ns=not significant.
AHR activation did not affect Wnt/β-catenin pathway gene expression
Albeit it is an inducible transcription factor, the AHR has a basal activation level through ligands
present in
normal media (tryptophan derivatives). In vivo, dietary and microbial ligands, and UVB-generated
ligands,
especially the tryptophan dimer FICZ, are relevant for the skin. Considering (i) that SSc manifests in
the
skin
very often and (ii) the results described above, we asked how AHR activation might affect the expression
of
Wnt/β-catenin pathway genes. We therefore incubated HDF cells with FICZ and determined the expression
levels of
the Wnt/β-catenin pathway genes (Fig. 1A). Surprisingly, the addition of FICZ to HDF cells had no
significant
effects, in contrast to the marked changes observed in AHR-deficient HDF (both shown in Fig. 1A). Thus,
ligand-mediated AHR activation did not alter the expression levels of the selected Wnt/β-catenin pathway
genes
in either HDF or HaCaT cells (Fig. 1A and 1B).
TGFβ effects on Wnt/β-catenin genes and β-catenin in AHR-deficient or -proficient HDF and HaCaT
cells
The addition of TGFβ to cultured fibroblasts is a recognized, simple fibrosis model [8], and canonical
Wnt/β-catenin signalling can be necessary for TGFβ-mediated fibrosis [34]. We therefore asked if the
Wnt/β-catenin pathway genes are modulated not only by AHR, but also by TGFβ in HDF and HaCaT cells.
Hence,
we
added TGFβ (10 ng/ml or 25 ng/ml) to the cultivated cells for up to 48 hours, and afterwards performed
ddPCR for
the Wnt/β-catenin pathway genes mentioned above. We also measured AHR expression itself to
determine if
it is changed by TGFβ addition. In HDF cells, with the exception of FZD2, addition of TGFβ to the
culture
medium reduced expression levels compared to the control (Fig. 2A). In the HDF-KO cells, significantly
more
FZD2 and CTNNB1 mRNAs were detectable after TGFβ exposure, but only at the higher TGFβ
dose
(Fig.
2B). In HaCaT cells, TGFβ addition did not affect any of the pathway genes, except for an increased
expression
of CTNNB1 (Fig. 2C). In HaCaT-KO cells, loss of AHR resulted in lower expression of FZD2 and
β-catenin
genes when TGFβ was added to cultures, even lower than the WT/KO decrease (Fig. 2D). The key observation
is that
TGFβ treatment led to opposite effects on CTNNB1 expression in the two skin cell types: it
decreased
CTNNB1 mRNA levels in HDF WT cells, while increasing them in HaCaT cells. Curiously, in HaCaT-KO
cells,
the combination “loss of AHR plus TGFβ addition” resulted in reduced Wnt/β-catenin pathway gene
expression.
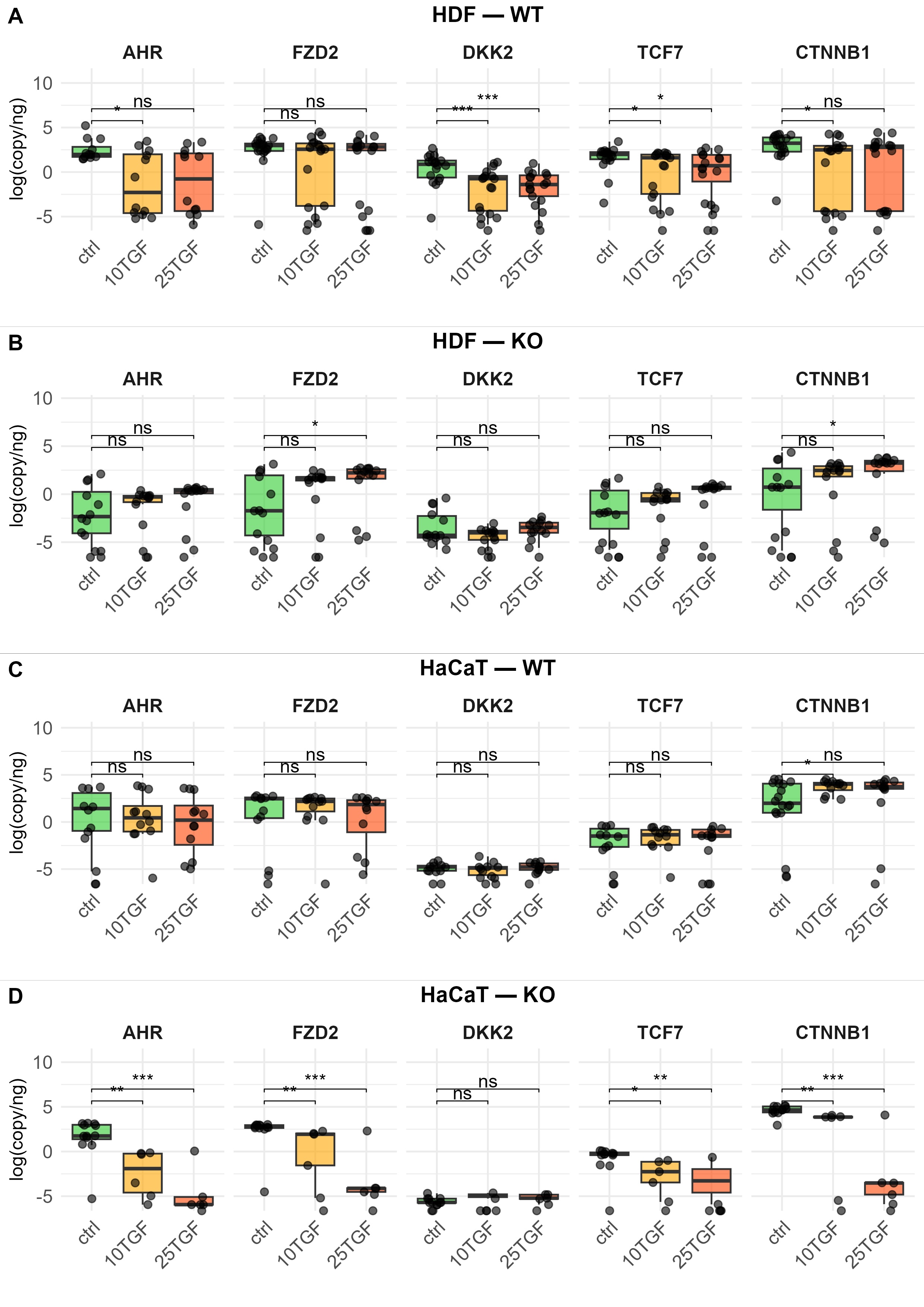
Fig. 2: Effect of TGFβ treatment on the expression of selected Wnt/β-catenin pathway genes, measured by ddPCR, in “wild-type” (WT) and AHR-deficient (KO) human dermal fibroblasts (HDF, panels A and B) and HaCaT (panels C and D). Cells were treated with TGFβ at 10 ng/mL (10TGF) or 25 ng/mL (25TGF) for 24 h and 48 h. Cells were collected from three independent experiments. Gene expression results from different timepoints were pooled and presented as log-transformed copies/ng. Boxplots with individual data points are shown. Control samples are shown in light green, while TGFβ-treated samples are shown in yellow (10TGF) and orange (25TGF). All samples were performed in triplicate. Statistical comparisons were performed using the Wilcoxon test. *: p <= 0.05; **: p <=0.01; ***: p <=0.001; ****: p <=0.0001; ns=not significant.
AHR deficiency in HDF abrogates TGFβ-induced collagen production in monocultures, as does the
presence
of
HaCaT cells in co-cultures
Collagen 1A1 (COL1A1), one of the main components of the ECM,
often dysregulated in fibrosis, is inducible upon TGFβ treatment in fibroblast culture [35]. We
treated
our
cells with TGFβ and assessed COL1A1 protein production by ELISA. As expected, HDF cells secreted more
COL1A1
upon TGFβ treatment (Fig. 3A, orange bars), already at 24 hours (with 25 ng/ml) and then further at 48
hours
later (detectable for both 10 ng/ml and 25 ng/ml TGFβ). In contrast, in HDF-KO cells, COL1A1 secretion
was
lower
than in HDF (about 50%), and only a very moderate increase was detectable after TGFβ addition (Fig.
3A,
orange
bars). Surprisingly, addition of FICZ inhibited TGFβ-mediated collagen production in HDF cells, and
somewhat
enhanced it in HDF-KO cells, especially at 48 hours. Nonetheless, absolute levels remained lower than
in
control
HDF or HDF-KO cells (Fig. 3A, green bars). Thus, in HDF cells, collagen production regulation involves
AHR
signaling as well as TGFβ signaling. Given the evidence in the literature that keratinocytes produce
factors
which influence collagen production by fibroblasts [3, 36], we next asked whether and how co-culturing
HaCaT
with HDF could affect collagen production by the latter (Fig. 3B). HaCaT were placed in the upper
chamber
inserted into a well and HDF were placed at the bottom of the well, so that soluble products (less
than
1000
kDa) could reach the lower well. As shown in Fig. 3B, co-culturing AHR-proficient HaCaT and HDF
(labeled
WT
co-culture in the figure) for 48 hours resulted in a collagen production of 2741.85 ± 7.46 ng/ml in
the
medium.
Addition of TGFβ to medium decreased collagen significantly by nearly 50%, to about 1465.38 ± 137.85
ng/ml. When
AHR was absent in both cell types (labelled as KO co-culture), the co-cultures´ supernatants yielded
lower
collagen than in co-cultured WT cells. This likely reflects that HDF-KO are low in collagen production
anyway
(Fig. 3A), and the presence of HaCaT-KO cells could not rescue this. Importantly, co-cultivation of
HaCaT
cells
with HDF cells (whether WT/WT or KO/KO) suppressed collagen production by TGFβ (presumably by the HDF
cells),
but otherwise presence or absence of AHR in the co-cultured HaCaT did not positively or negatively
influence
collagen production.
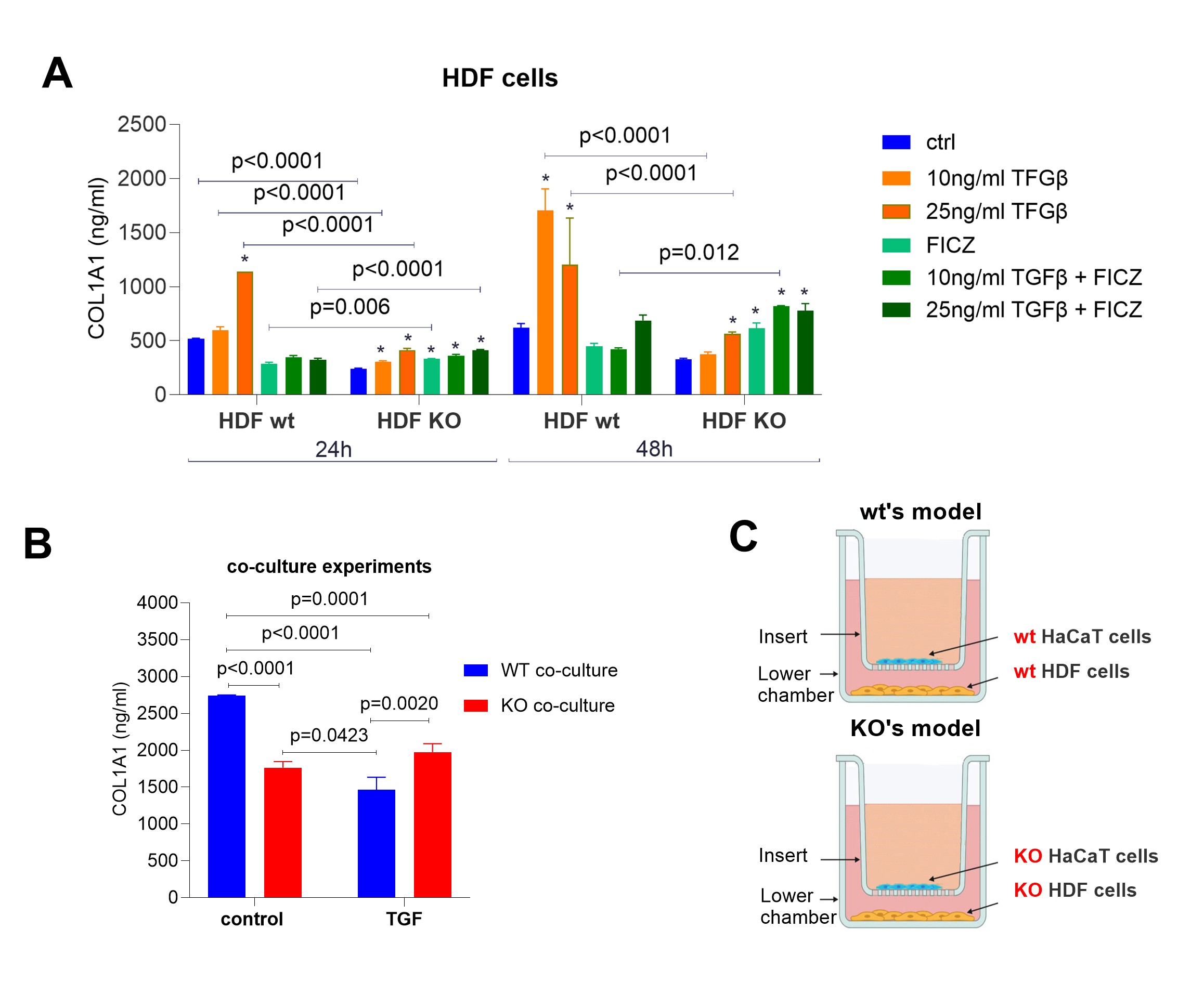
Fig. 3: COL1A1 secretion (ng/ml) by HDF cells (A) and HDF-HaCaT co-culture (B). (A) COL1A1 concentrations in HDF and HDF-KO cells after treatment with TGFβ (10 ng/ml, 25 ng/ml), FICZ, and their combinations for 24 and 48 hours. Control (untreated) samples are also shown. (B) COL1A1 secretion in co-cultures of HDF and HaCaT cells (wild-type (wt) and KO) after treatment with TGFβ (25 ng/ml) for 48 hours.(C) Schematic representation of the co-culture setup: the “wild-type” WT model consists of “wild-type” HDF and HaCaT cells, while the KO model includes AHR-deficient HDF and HaCaT cells. HDF cells were seeded in the lower chamber, and HaCaT cells were cultured on an insert. All samples were performed in triplicate. Two-way ANOVA. Bar plot. Mean ± SD. Post hoc multiple comparisons. p ≤ 0.05. * indicates statistically significant difference in comparison to the corresponding untreated control.
MMP1 secretion by TGFβ addition is dependent on AHR
MMP1 production is important for degrading collagen and thus regulating ECM. Excessive collagen
deposition
due
to a lack of degradation significantly adds to the fibrosis process. HDF cells produced robust amounts
of
MMP1,
but TGFβ-treatment almost halved this over 24/48 hours of cultivation (Fig. 4A), and MMP1 production
was
entirely lost in AHR-deficient HDF. Highlighting the role of AHR in this, the treatment of HDF with
FICZ
rescued
the TGFβ-mediated down-regulation and HDF-KO almost stopped secreting MMP1 upon FICZ addition.
Although
keratinocytes were described to produce MMP1 as well [37], in our hands HaCaT cells were very low
producers, and
TGFβ addition lowered secretion even further. AHR-deficient HaCaT were non-producers (Fig. 4B). We
conclude that
HaCaT cells contribute much less to MMP1-mediated collagen degradation than fibroblasts. Co-culturing
HaCaT and
HDF (both AHR-proficient) did not lessen the TGFβ-induced decrease of MMP1 production (Fig. 4C compare
Fig. 4A
and 4B). Congruent with the data of mono-cultures, MMP1 production was almost entirely lost in HDF KO
cells, and
the presence of HaCaT cells in the cultures did not change this. Taken together, AHR plays a crucial
role
in
MMP1 production by HDF.
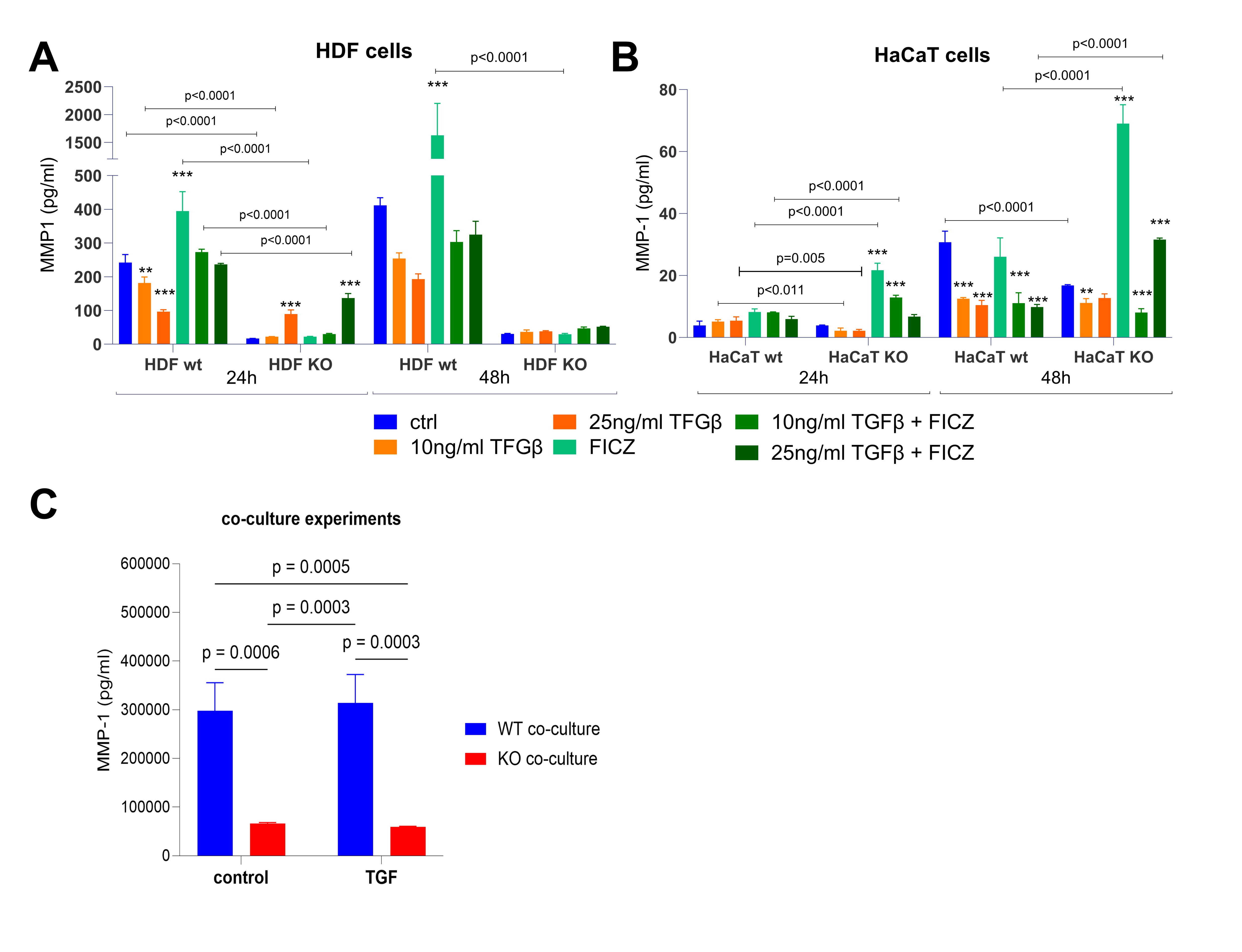
Fig. 4: MMP-1 secretion (pg/ml) in HDF and HaCaT cells cultured in monoculture (A, B) and co-culture (C). (A) MMP-1 concentrations in HaCaT WT and AHR KO cells after treatment with TGFβ (10 ng/ml, 25 ng/ml), FICZ, and their combinations for 24 and 48 hours. Control (untreated) samples are also shown. (B) MMP-1 concentrations in HDF WT and AHR KO cells under the same conditions as in (A). (C) MMP-1 secretion in co-culture of HDF and HaCaT (WT and AHR KO) after treatment with TGFβ (25 ng/ml) for 48 hours. HDF cells were seeded at the bottom of the well, while HaCaT cells were cultured on the inserts, allowing indirect interaction between the two cell types. All samples were performed in triplicate. Two-way ANOVA. Bar plot. Mean ± SD. Post hoc multiple comparisons. p ≤ 0.05.
Proteome profiler in co-culture
So far, we identified AHR to be involved in the expression/production of β-catenin,
collagen, and MMP1, in both mono- and co-cultures. To further unravel the role of AHR, we next used a
proteome
profiler to compare protein secretion in the supernatants of WT/WT and KO/KO co-cultures
(Supplementary
material
Table S2). We observed that vitamin D binding protein (vitamin D BP), retinol-binding protein 4
(RBP-4),
platelet factor 4 (PF4, CXCL4), angiogenin, and adiponectin were the most upregulated proteins,
whereas
IL-8
(CXCL8) and monocyte chemoattractant protein-1 (MCP-1) exhibited the most pronounced downregulation in
the
KO/KO
co-culture model compared to WT/WT (Fig. 5). To validate the downregulation of IL-8 observed in the
proteome
profiler, we performed an independent ELISA assay, which confirmed a significantly reduced IL-8
secretion
in
KO/KO co-cultures (Figure S6, Supplementary material). The analyzed proteome profiler indicated a loss
of
specific protein signals in KO co-cultures within pathways related to rheumatoid arthritis,
cytokine-receptor
interactions, and TNF signaling. These alterations in signaling pathways between the experimental
co-culture
models highlight the impact of AHR on inflammatory processes and immune-related signaling cascades.
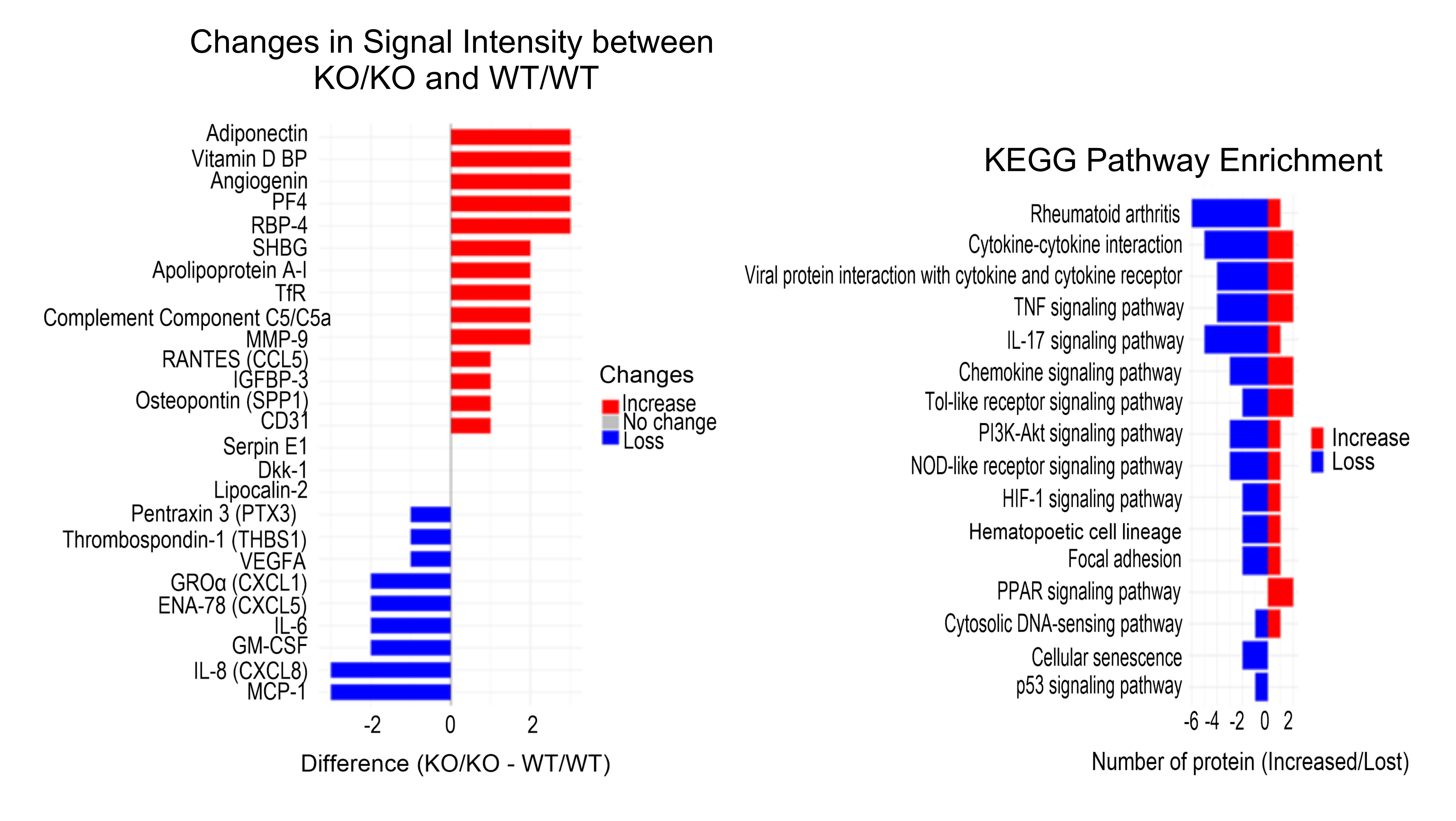
Fig. 5: Differential protein expression and KEGG pathway enrichment between KO/KO and WT/WT cell cultures. Left panel - changes in protein signal intensity between KO/KO and WT/WT groups measured in the supernatant of cell cultures. Signal intensity was visually assessed and semi-quantitatively scored on a 4-point scale: 0 = no expression, 1 = weak (+), 2 = moderate (++), 3 = strong (+++). The x-axis shows the difference in signal (KO/KO – WT/WT), with positive values (blue) indicating increased signal and negative values (red) indicating decreased or absent signal in KO/KO cells. Red bars indicate upregulated proteins, while blue bars represent downregulated/loss proteins in the KO/KO group relative to WT/WT. Right panel - KEGG pathway enrichment analysis. The x-axis represents the number of proteins per pathway that showed increased (red) or decreased (blue) signal in KO/KO compared to WT/WT samples, highlighting the biological pathways most affected by AHR deletion.
AHR deficiency impaired cells motility
Impaired wound healing can be found in SSc and a majority of patients experience digital ulceration at
some
point. We therefore assessed the relevance of AHR for the ability of HaCaT and HDF cells to close a
gap
in
full
cell layers, as an albeit limited token of two fibrosis-relevant functions: cell migration and wound
healing. We
introduced a gap with a pipette tip and microscopically quantified the area closed by immigrating
cells
20/30
hours (HDF) and 24/48 hours (HaCaT) later. Proliferation was not specifically quantified, so its
contribution
cannot be considered separately. Fig. 6A-C shows the results for HaCaT, HaCaT-KO, HDF, or HDF cells
treated with
a chemical inhibitor of AHR signaling, CH223191, following treatment with TGFβ, FICZ, or a combination
of
both.
While HaCaT cells were capable of slowly closing this “wound”, the addition of FICZ or TGFβ (or both)
vastly
enhanced this process, with FICZ plus TGFβ resulting in full closure 48 hours after scratching (Fig.
6A).
In
contrast, HaCaT-KO were very slow in re-filling the scratch area, and only TGFβ addition (or TGFβ plus
FICZ),
but not FICZ alone, supported full closure (Fig. 6B, 6D). Thus, AHR-mediated TGFβ signaling is a major
factor in
the gap-closing capacity of HaCaT cells, and our data with HaCaT-KO/HDF plus chemical inhibition of
AHR
activity
suggests that AHR signaling modulates TGFβ-induced changes in cell migration.
As expected, the scratch wound assay revealed that HDF are much better at migration than HaCaT cells
(Fig.
6E),
indeed, the scratch had closed at least 70% by 48 hours (compared to 30% in HaCaT). Again, the
addition
of
FICZ
and/or TGFβ accelerated the migration, while inhibition of AHR signaling slowed it down.
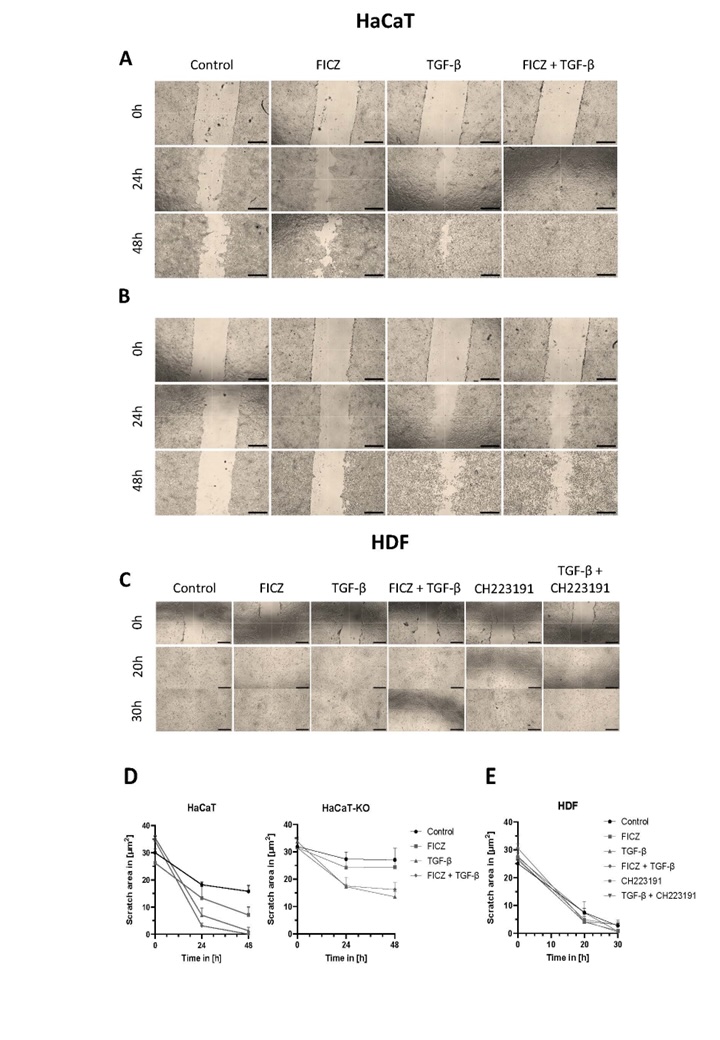
Fig. 6: Cell migration during wound healing in HaCaT (A), HaCaT-KO (B), and HDF (C) cells. Representative transmitted light microscopy images from scratch assays performed using a 200 µL pipette tip. Cells were treated with TGFβ (25 ng/mL), FICZ (100 nM), or a combination of both. For HDF cells (C), additional conditions included treatment with the AHR inhibitor CH223191 (10 µM), either alone or in combination with TGFβ. Images were acquired at 0, 24, and 30 or 48 hours post-scratch. Quantitative analysis of wound closure over time is shown for HaCaT and HaCaT-KO cells in panel D, and for HDF cells in panel E; data are presented as mean ± standard deviation (s.d.). Scale: 40× magnification; scale bar, 500 µm.
Discussion
Systemic sclerosis (SSc) is characterized by dysregulated collagen synthesis and degradation, accompanied by aberrant immune signaling. Important signaling pathways are TGFβ and/or Wnt/β-catenin. Here, we present data indicating that the aryl hydrocarbon receptor (AHR) is also involved in these processes. In this study, we employed (i) a simple fibrosis model by adding TGFβ to HDF and (ii) used AHR-deficient HDF and HaCaT cells to assess a possible additional impact of the AHR. Our study showed that the AHR had a significant role in maintaining cell functionality, including the response to fibrotic agents, particularly TGFβ, and collagen production.
Wnt/β-catenin and AHR
Regarding Wnt/β-catenin signaling, an important player in SSc, we identified an influence of the AHR
on
β-catenin mRNA expression as well as on genes of its upstream signaling pathway. In AHR-deficient HDF
cells, we
observed lower levels of β-catenin mRNA compared to AHR-proficient cells, whereas in HaCaT cells, the
opposite
trend was observed. Given the known cell-specificity of the AHR, this finding was not unexpected.
While
recent
studies have highlighted potential crosstalk between the AHR and Wnt/β-catenin signaling, no curated
pathways in
professional academic databases currently establish a direct mechanistic links between AHR,
Wnt/β-catenin,
(or
TGFβ) signaling. One study showed by co-immunoprecipitation and immunofluorescence assays that the AHR
associates with E-cadherin and β-catenin in normal murine mammary gland cells (NMuMG), suggesting its
involvement in cell-cell adhesion [38]. Our study provided preliminary evidence indicating that
AHR
status significantly influences the regulation of the Wnt/β-catenin pathway in a cell type-dependent
manner (HDF
vs. HaCaT). Our observations align with the evidence that β-catenin functions primarily in
transcriptional
regulation in fibroblasts but is sequestered at adherens junctions in keratinocytes [39]. Available
studies
generally describe that the absence of AHR is associated with increased activity of the Wnt/β-catenin
pathway,
including elevated β-catenin levels [26–28, 40]. While this trend is observed in many tissues,
exceptions
do
exist [41, 42], suggesting a more nuanced regulation. In our work, we observed such an increased Wnt
activity
only in HaCaT cells, but not in HDF. Of note, AHR’s ability to modulate β-catenin levels extends
beyond
transcriptional control — acting as an E3 ubiquitin ligase, it can directly facilitate β-catenin
degradation in
a ligand-dependent manner, requiring molecules such as tryptophan or glucosinolates [43]. In
fibroblasts,
AHR
may enhance, support, or maintain the Wnt/β-catenin pathway, whereas in keratinocytes it may restrain
its
expression. This may be due to the different role of the receptor in these cell types. Therefore, the
mechanistic relationship between AHR and Wnt/β-catenin signaling is cell- and context-dependent and
apparently
does not appear to result solely from classical AHR activation through ligands and transcriptional
regulation,
suggesting post-translational regulation, protein-protein interaction, or impact on the stabilization
of
other
pathway regulators. Moreover, in our hands, HDF-KO cells exhibited decreased transcription of analyzed
Wnt/β-catenin pathway genes following TGFβ treatment, whereas HDF cells showed increased expression.
This
finding may support the anti-fibrotic role of the feedback loop between AHR and TGFβ.
At the same time, our combination experiments with TGFβ addition on wild-type versus AHR-deficient HDF
cells
revealed that collagen production triggered by TGFβ needs the AHR to reach optimal levels in these
fibroblasts.
TGFβ and AHR – reciprocal regulation
Fibrosis is a natural part of the healing process, but under pathological conditions such as SSc, it
becomes a
complex phenomenon that reduces patients’ quality of life, often causing significant pain and
increasing
mortality rates. In SSc, the extent of skin involvement correlates with a worse prognosis [44].
Although
the AHR
has been investigated in fibrosis, findings remain inconclusive or even contradictory [18, 45–49]. The
AHR
is
expressed in all skin cells [50] and is well known for its cell-specificity and manifold, sometimes
contradictory roles in regulating cellular functions [51, 52]. The AHR-TGFβ relationship is complex,
as
fibroblasts lacking the AHR secrete higher levels of TGFβ [53], whereas AHR activation is associated
with
reduced fibrotic responses [2, 54]. However, some inconsistencies have also been reported,
particularly
in
intestinal studies [49, 55]. TGFβ downregulates AHR transcription, while the AHR modulates TGFβ
signaling
through interactions with key regulatory proteins such as latent TGFβ-binding protein (LTBP1) and
SMADs.
This
reciprocal regulation highlights the complexity of the AHR-TGFβ interplay in fibrotic processes [56,
57].
It has
long been known that the outcome of AHR activity is cell specific, and our data underscore this again.
In
HDF
and HaCaT cells, removal of AHR or activation of AHR via the high-affinity ligand FICZ had different
outcomes,
as did the addition of TGFβ.
TGFβ and AHR – crosstalk in HDF and HaCaT
Under standard monoculture conditions, treatment of skin fibroblasts with TGFβ typically induces an
increase in
type I collagen secretion [35]. Interestingly, the co-culture of HDF and HaCaT resulted in lower
collagen
(COL1A1) production upon TGFβ treatment. This finding strongly suggests crosstalk between these two
main
skin-cell types of the epidermis and dermis. This clearly may have an impact on cellular functions,
such
as
collagen production, cytokine secretion, and gene expression, as well as regulatory mechanisms under
fibrotic
conditions, which is consistent with the literature [4, 36].
Our results also confirmed that loss of the AHR in HDF abolished MMP1 secretion (Fig. 4), consistent
with
previous studies [7, 18], which demonstrated that AHR is an indispensable regulator of matrix
degradation.
Given
that excessive collagen accumulation due to impaired MMP1 activity is a hallmark of fibrosis, our
findings
highlight the potential of the AHR as a therapeutic target for restoring ECM balance.
Cytokines
Moreover, in the co-culture of AHR-deficient cells, we observed disruptions and reduced signaling of
proteins
associated with rheumatoid arthritis, as well as cytokine interactions involving TNF and IL-17. An
active
AHR
typically targets several genes, either directly or via a cascade triggered by those direct targets.
As
expected, proteome analysis identified several pathways, with the KEGG database indicating mainly
inflammation-related events. This is consistent with the known role of AHR in immune balance. This
sensitivity
can be clinically important, as many SSc patients have low levels of AHR expression in skin
fibroblasts.
We
observed that AHR-KO cells in co-culture secreted neither monocyte chemoattractant protein-1 (MCP-1)
nor
IL-8,
both of which typically may be produced by keratinocytes and fibroblasts [58]. MCP-1 plays a role in
inducing
cellular senescence in keratinocytes [59] and stimulates inflammatory cells to the skin [60], whereas
IL-8
may
promote keratinocyte migration, which is important for wound healing [61]. While more research is
needed,
these
data underscore the importance of the AHR in fibroblasts and their fibrogenic capacity.
Migration
A study using mouse skin tissue explants demonstrated that the lack of AHR accelerated skin
re-epithelialization
by enhancing keratinocyte migration, without affecting cell proliferation or the recruitment of
inflammatory
cells [62]. In contrast to this, we observed that AHR-deficient HaCaT cells were strongly impaired in
closing
the scratch gap within two days, and neither TGFβ nor FICZ addition helped. HaCaT cells were able to
close
the
scratch gap, but HDF cells performed even better and faster (while chemical inhibition of
AHR-signaling
slowed
this down). In both cell types, the addition of FICZ and/or TGFβ accelerated this
process.
Note, however, that such scratch assay does not allow for differentiation between cell migration and
proliferation. Additional functional tests, such as proliferation markers or time-lapse imaging, will
be
necessary to determine the exact mechanisms of this effect.
Immune cells
The proteome profiling data showed that immune-related proteins were modulated in the co-cultures.
Keratinocytes
are immune cells in the sense that they produce cytokines in response to injury, several of which were
identified in the co-culture proteome profile (e.g. CXCL8 (IL8), MCP-1 (a monocyte attractant), or
CCL5
(RANTES)
- a T cell/eosinophil attractant. SSc is an autoimmune disease, thus immunological events are
important.
Due to
the exploratory and rather qualitative nature of the proteome profiling, these observations should be
interpreted as hypothesis generating, but are biologically consistent with the known function of
keratinocytes
and fibroblasts. More research is warranted on the role of the AHR in this context, i.e., the highly
dynamic
interactions between fibroblasts, keratinocytes, and immune cells. Therefore, future studies should
incorporate
immune components and address their involvement [63].
Conclusion
In conclusion, AHR-signaling is part of a three-directional crosstalk between AHR-TGFβ-Wnt/β-catenin in human dermal fibroblasts. This crosstalk was weaker or did not exist in HaCaT cells. Nonetheless, our co-cultivation experiments demonstrate a contribution of HaCaT cells to the fibrogenic process.
Our findings underscore the multifaceted, context-dependent nature of the AHR in fibrosis and skin homeostasis, highlighting distinct roles in fibroblasts and keratinocytes. Specifically, in fibroblasts, AHR appears crucial for balancing ECM turnover, whereas in keratinocytes, its role may be more structural. Consequently, rather than posing a simple “on/off” question regarding AHR, these results suggest that any therapeutic strategy may lie in the fine-tuning of AHR activity at the level of specific cell types—a challenging proposition, given the complexity of fibrotic processes and the diverse roles the AHR plays in different cellular environments.
For future studies, more refined co-culture models, including the incorporation of immune and endothelial cells, are important to examine the cross-talk between fibroblasts and keratinocytes. Such multifaceted models will provide a more comprehensive view of how AHR modulation might be therapeutically leveraged in systemic sclerosis and related fibrotic conditions.
Abbreviations
AHR aryl hydrocarbon receptor; ECM extracellular matrix; FICZ 6-formylindolo [3, 2-b]carbazole; HDF human dermal fibroblasts; KO knock-out; MMP1 matrix metalloproteinase 1; SSc Systemic sclerosis; TGFβ transforming growth factor beta; WT wild-type;
Acknowledgements
We thank Professor Thomas Krieg and Doctor Beate Eckes from the University of Cologne for their valuable scientific discussion, which provided important perspectives and helped refine our interpretation of the findings. We are grateful to our respective institutes for their hospitality in hosting the exchange researchers. We thank Yana Kaliberda and Peter Jacobi for contributions to preliminary experiments and Alexander Meißner for help with Fig. 6.
Author Contributions
Conceptualization – A.W., C.E.; methodology – A.W., A. E-M, D.H.; formal analysis- B.S, A.W.;
investigation- A.
E-M, D.H, G.F., I.H, A.W.; generation of CRISPR-modified cell lines – A.R.; writing – original draft
preparation
- A.W., C.E.; writing—review and editing, A.G-P, T.H-S., C.E.
Funding Sources
This work was co-funded by the Deutscher Akademischer Austauschdienst (DAAD, project 57655777), the
Polish
National Agency for Academic Exchange (NAWA, BPN/BDE/2022/1/00010), the Deutsche
Forschungsgemeinschaft
(FOR5489), and the Polish Ministry of Science and Higher Education through statutory funding for the
National
Institute of Geriatrics, Rheumatology and Rehabilitation (statutory grant no. S15).
Statement of Ethics
The authors have no ethical conflicts to disclose. This study did not involve human participants or
experiments
on animals.
Disclosure Statement
The authors have no conflicts of interest to declare.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work the authors used Grammarly and ChatGPT in order to improve the
readability
and language of the manuscript.
References
| 1 | Lehmann GM, Xi X, Kulkarni AA, Olsen KC, Pollock SJ, Baglole CJ, Gupta S, Casey AE, Huxlin KR,
Sime PJ, Feldon SE, Phipps RP. The aryl hydrocarbon receptor ligand ITE inhibits TGFβ1-induced
human
myofibroblast differentiation. Am J Pathol. 2011;178(4):1556-67.
https://doi.org/10.1016/j.ajpath.2010.12.025 |
| 2 | Woeller CF, Roztocil E, Hammond CL, Feldon SE, Phipps RP. The Aryl Hydrocarbon Receptor and
Its
Ligands Inhibit Myofibroblast Formation and Activation: Implications for Thyroid Eye Disease. Am
J
Pathol. 2016;186(12):3189-202.
https://doi.org/10.1016/j.ajpath.2016.08.017 |
| 3 | Maas-Szabowski N, Shimotoyodome A, Fusenig NE. Keratinocyte growth regulation in fibroblast
cocultures via a double paracrine mechanism. J Cell Sci. 1999;112(12):1843-53.
https://doi.org/10.1242/jcs.112.12.1843 |
| 4 | Garner WL. Epidermal Regulation of Dermal Fibroblast Activity. Plast Reconstr Surg.
1998;102(1):135-9.
https://doi.org/10.1097/00006534-199807000-00021 |
| 5 | Russo B, Brembilla NC, Chizzolini C. Interplay between keratinocytes and fibroblasts: A
systematic
review providing a new angle for understanding skin fibrotic disorders. Front Immunol.
2020;6;11:648.
https://doi.org/10.3389/fimmu.2020.00648 |
| 6 | Leask A. Getting Out of a Sticky Situation: Targeting the Myofibroblast in Scleroderma. Open
Rheumatol J. 2012;6:163-9.
https://doi.org/10.2174/1874312901206010163 |
| 7 | Murai M, Yamamura K, Hashimoto-Hachiya A, Tsuji G, Furue M, Mitoma C. Tryptophan photo-product
FICZ upregulates AHR/MEK/ERK-mediated MMP1 expression: Implications in anti-fibrotic
phototherapy.
J
Dermatol Sci. 2018;91(1):97-103.
https://doi.org/10.1016/j.jdermsci.2018.04.010 |
| 8 | Xu Q, Norman JT, Shrivastav S, Lucio-Cazana J, Kopp JB. In vitro models of TGF-β-induced
fibrosis
suitable for high-throughput screening of antifibrotic agents. Am J Physiol Renal Physiol.
2007;293(2):F631-40.
https://doi.org/10.1152/ajprenal.00379.2006 |
| 9 | Schneider AJ, Branam AM, Peterson RE. Intersection of AHR and Wnt signaling in development,
health, and disease. Int J Mol Sci. 2014;3;15(10):17852-85.
https://doi.org/10.3390/ijms151017852 |
| 10 | Duspara K, Bojanic K, Pejic JI, Kuna L, Kolaric TO, Nincevic V, Smolic R, Vcev A, Glasnovic M,
Curcic IB, Smolic M. Targeting the Wnt Signaling Pathway in Liver Fibrosis for Drug Options: An
Update. J Clin Transl Hepatol. 2021;9(6):960-71.
https://doi.org/10.14218/JCTH.2021.00065 |
| 11 | Hamburg-Shields E, Dinuoscio GJ, Mullin NK, Lafayatis R, Atit RP. Sustained β-catenin activity
in
dermal fibroblasts promotes fibrosis by up-regulating expression of extracellular matrix
protein-coding genes. J Pathol. 2015;235(5):686-97.
https://doi.org/10.1002/path.4481 |
| 12 | Lam AP, Flozak AS, Russell S, Wei J, Jain M, Mutlu GM, Budinger GR, Feghali-Bostwick CA, Varga
J,
Gottardi CJ. Nuclear β-Catenin Is Increased in Systemic Sclerosis Pulmonary Fibrosis and
Promotes
Lung Fibroblast Migration and Proliferation. Am J Respir Cell Mol Biol. 2011;45(5):915-22.
https://doi.org/10.1165/rcmb.2010-0113OC |
| 13 | Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X, Zhou Z, Shu G, Yin G. Wnt/β-catenin signalling:
function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther.
2022;7(1):3.
https://doi.org/10.1038/s41392-021-00762-6 |
| 14 | Wei J, Fang F, Lam AP, Sargent JL, Hamburg E, Hinchcliff ME, Gottardi CJ, Atit R, Whitfield
ML,
Varga J. Wnt/β-catenin signaling is hyperactivated in systemic sclerosis and induces
Smad-dependent
fibrotic responses in mesenchymal cells. Arthritis Rheum. 2012;64(8):2734-45.
https://doi.org/10.1002/art.34424 |
| 15 | Haarmann-Stemmann T, Esser C, Krutmann J. The Janus-Faced Role of Aryl Hydrocarbon Receptor
Signaling in the Skin: Consequences for Prevention and Treatment of Skin Disorders. J Invest
Dermatol. 2015;135(11):2572-6.
https://doi.org/10.1038/jid.2015.285 |
| 16 | Esser C, Bargen I, Weighardt H, Haarmann-Stemmann T, Krutmann J. Functions of the aryl
hydrocarbon
receptor in the skin. Semin Immunopathol. 2013;35(6):677-91.
https://doi.org/10.1007/s00281-013-0394-4 |
| 17 | Shi Y, Tang B, Yu J, Luo Y, Xiao Y, Pi Z, Tang R, Wang Y, Kanekura T, Zeng Z, Xiao R. Aryl
hydrocarbon receptor signaling activation in systemic sclerosis attenuates collagen production
and
is a potential antifibrotic target. Int Immunopharmacol. 2020;88:106886.
https://doi.org/10.1016/j.intimp.2020.106886 |
| 18 | Roztocil E, Hammond CL, Gonzalez MO, Feldon SE, Woeller CF. The aryl hydrocarbon receptor
pathway
controls matrix metalloproteinase-1 and collagen levels in human orbital fibroblasts. Sci Rep.
2020;10(1):8477.
https://doi.org/10.1038/s41598-020-65414-1 |
| 19 | Abdelrahman MA, Sakr HM, Shaaban MAA, Afifi N. Serum and synovial matrix metalloproteinases 1
and
3 in patients with early rheumatoid arthritis: potentially prospective biomarkers of
ultrasonographic joint damage and disease activity. Egypt J Intern Med. 2019;31(4):965-71.
https://doi.org/10.4103/ejim.ejim_163_19 |
| 20 | Frost J, Ramsay M, Mia R, Moosa L, Musenge E, Tikly M. Differential gene expression of MMP-1,
TIMP-1 and HGF in clinically involved and uninvolved skin in South Africans with SSc.
Rheumatology.
2012;51(6):1049-52.
https://doi.org/10.1093/rheumatology/ker367 |
| 21 | Sadeghi Shaker M, Rokni M, Kavosi H, Enayati S, Madreseh E, Mahmoudi M, Farhadi E, Vodjgani M.
Salirasib Inhibits the Expression of Genes Involved in Fibrosis in Fibroblasts of Systemic
Sclerosis
Patients. Immun Inflamm Dis. 2024;12(11):e70063.
https://doi.org/10.1002/iid3.70063 |
| 22 | Yang Y, Chan WK. Glycogen Synthase Kinase 3 Beta Regulates the Human Aryl Hydrocarbon Receptor
Cellular Content and Activity. Int J Mol Sci. 2021;22(11):6097.
https://doi.org/10.3390/ijms22116097 |
| 23 | Yang Y, Tao Y, Yi X, Zhong G, Gu Y, Cui Y, Zhang Y. Crosstalk between aryl hydrocarbon
receptor
and Wnt/β-catenin signaling pathway: Possible culprit of di (2-ethylhexyl) phthalate-mediated
cardiotoxicity in zebrafish larvae. Sci Total Environ. 2024;907:167907.
https://doi.org/10.1016/j.scitotenv.2023.167907 |
| 24 | Baljinnyam B, Klauzinska M, Saffo S, Callahan R, Rubin JS. Recombinant R-spondin2 and Wnt3a
Up-
and Down-Regulate Novel Target Genes in C57MG Mouse Mammary Epithelial Cells. PLoS One.
2012;7(1):e29455.
https://doi.org/10.1371/journal.pone.0029455 |
| 25 | Li CH, Liu CW, Tsai CH, Peng YJ, Yang YH, Liao PL, Lee CC, Cheng YW, Kang JJ. Cytoplasmic aryl
hydrocarbon receptor regulates glycogen synthase kinase 3 beta, accelerates vimentin
degradation,
and suppresses epithelial-mesenchymal transition in non-small cell lung cancer cells. Arch
Toxicol.
2017;91(5):2165-78.
https://doi.org/10.1007/s00204-016-1870-0 |
| 26 | Zhang H, Yao Y, Chen Y, Yue C, Chen J, Tong J, Jiang Y, Chen T. Crosstalk between AhR and
wnt/β-catenin signal pathways in the cardiac developmental toxicity of PM2.5 in zebrafish
embryos.
Toxicology. 2016;355-356:31-8.
https://doi.org/10.1016/j.tox.2016.05.014 |
| 27 | Tong Y, Niu M, Du Y, Mei W, Cao W, Dou Y, Yu H, Du X, Yuan H, Zhao W. Aryl hydrocarbon
receptor
suppresses the osteogenesis of mesenchymal stem cells in collagen-induced arthritic mice through
the
inhibition of β-catenin. Exp Cell Res. 2017;350(2):349-57.
https://doi.org/10.1016/j.yexcr.2016.12.009 |
| 28 | Procházková J, Kabátková M, Bryja V, Umannová L, Bernatík O, Kozubík A, Machala M, Vondrácek
J.
The Interplay of the Aryl Hydrocarbon Receptor and β-Catenin Alters Both AhR-Dependent
Transcription
and Wnt/β-Catenin Signaling in Liver Progenitors. Toxicol Sci. 2011;122(2):349-60.
https://doi.org/10.1093/toxsci/kfr129 |
| 29 | Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal
keratinization
in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol.
1988;106(3):761-71.
https://doi.org/10.1083/jcb.106.3.761 |
| 30 | Vogeley C, Sondermann NC, Woeste S, Momin AA, Gilardino V, Hartung F, Heinen M, Maaß SK,
Mescher
M, Pollet M, Rolfes KM, Vogel CFA, Rossi A, Lang D, Arold ST, Nakamura M, Haarmann-Stemmann
T.Unraveling the differential impact of PAHs and dioxin-like compounds on AKR1C3 reveals the
EGFR
extracellular domain as a critical determinant of the AHR response. Environ Int.
2022;158:106989.
https://doi.org/10.1016/j.envint.2021.106989 |
| 31 | Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R Package for Comparing Biological Themes
Among
Gene Clusters. OMICS. 2012;16(5):284-7.
https://doi.org/10.1089/omi.2011.0118 |
| 32 | Carlson M: org.Hs.eg.db. Bioconductor Annotaion Package. 2023. DOI:
10.18129/B9.bioc.org.Hs.eg.db
|
| 33 | Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer International Publishing;
2016.
DOI: 10.1007/978-3-319-24277-4
https://doi.org/10.1007/978-3-319-24277-4 |
| 34 | Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, Horn A, Kireva T, Beyer C,
Zwerina J, Schneider H, Sadowski A, Riener MO, MacDougald OA, Distler O, Schett G, Distler JH.
Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun.
2012;3:735.
https://doi.org/10.1038/ncomms1734 |
| 35 | Juhl P, Bondesen S, Hawkins CL, Karsdal MA, Bay-Jensen AC, Davies MJ, Siebuhr AS. Dermal
fibroblasts have different extracellular matrix profiles induced by TGF-β, PDGF and IL-6 in a
model
for skin fibrosis. Sci Rep. 2020;10(1):17300.
https://doi.org/10.1038/s41598-020-74179-6 |
| 36 | Ghaffari A, Kilani RT, Ghahary A. Keratinocyte-Conditioned Media Regulate Collagen Expression
in
Dermal Fibroblasts. J Invest Dermatol. 2009;129(2):340-7.
https://doi.org/10.1038/jid.2008.253 |
| 37 | Tandara AA, Mustoe TA. MMP- and TIMP-secretion by human cutaneous keratinocytes and
fibroblasts
-
Impact of coculture and hydration. J Plast Reconstr Aesthet Surg. 2011;64(1):108-16.
https://doi.org/10.1016/j.bjps.2010.03.051 |
| 38 | Rico-Leo EM, Alvarez-Barrientos A, Fernandez-Salguero PM. Dioxin receptor expression inhibits
basal and transforming growth factor β-induced epithelial-to-mesenchymal transition. J Biol
Chem.
2013;288(11):7841-56.
https://doi.org/10.1074/jbc.M112.425009 |
| 39 | Söderholm S, Cantù C. The WNT/β-catenin dependent transcription: A tissue-specific business.
WIREs
Mech Dis. 2021;13(3):e1511.
https://doi.org/10.1002/wsbm.1511 |
| 40 | Wincent E, Stegeman JJ, Jönsson ME. Combination effects of AHR agonists and Wnt/β-catenin
modulators in zebrafish embryos: Implications for physiological and toxicological AHR functions.
Toxicol Appl Pharmacol. 2015;284(2):163-79.
https://doi.org/10.1016/j.taap.2015.02.014 |
| 41 | Shackleford G, Sampathkumar NK, Hichor M, Weill L, Meffre D, Juricek L, Laurendeau I,
Chevallier
A, Ortonne N, Larousserie F, Herbin M, Bièche I, Coumoul X, Beraneck M, Baulieu EE, Charbonnier
F,
Pasmant E, Massaad C. Involvement of Aryl hydrocarbon receptor in myelination and in human nerve
sheath tumorigenesis. Proc Natl Acad Sci U S A. 2018;115(6):E1319-28.
https://doi.org/10.1073/pnas.1715999115 |
| 42 | Moreno-Marín N, Merino JM, Alvarez-Barrientos A, Patel DP, Takahashi S, González-Sancho JM,
Gandolfo P, Rios RM, Muñoz A, Gonzalez FJ, Fernández-Salguero PM. Aryl Hydrocarbon Receptor
Promotes
Liver Polyploidization and Inhibits PI3K, ERK, and Wnt/β-Catenin Signaling. iScience.
2018;4:44-63.
https://doi.org/10.1016/j.isci.2018.05.006 |
| 43 | Kawajiri K, Kobayashi Y, Ohtake F, Ikuta T, Matsushima Y, Mimura J, Pettersson S, Pollenz RS,
Sakaki T, Hirokawa T, Akiyama T, Kurosumi M, Poellinger L, Kato S, Fujii-Kuriyama Y Proc Natl
Acad
Sci U S A. 2009;106(32):13481-6.
https://doi.org/10.1073/pnas.0902132106 |
| 44 | Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology
(Oxford). 2009;48 Suppl 3:iii14-8.
https://doi.org/10.1093/rheumatology/kep108 |
| 45 | Takei H, Yasuoka H, Yoshimoto K, Takeuchi T. Aryl hydrocarbon receptor signals attenuate lung
fibrosis in the bleomycin-induced mouse model for pulmonary fibrosis through increase of
regulatory
T cells. Arthritis Res Ther. 2020;22(1). DOI: 10.1186/S13075-020-2112-7
https://doi.org/10.1186/s13075-020-2112-7 |
| 46 | Liu Y, Zhao N, Xu Q, Deng F, Wang P, Dong L, Lu X, Xia L, Wang M, Chen Z, Zhou J, Zuo D. MBL
Binding with AhR Controls Th17 Immunity in Silicosis-Associated Lung Inflammation and Fibrosis.
J
Inflamm Res. 2022;15:4315-29.
https://doi.org/10.2147/JIR.S357453 |
| 47 | Wu SM, Tsai JJ, Pan HC, Arbiser JL, Elia L, Sheu ML. Aggravation of pulmonary fibrosis after
knocking down the aryl hydrocarbon receptor in the insulin-like growth factor 1 receptor
pathway.
Br
J Pharmacol. 2022;179(13):3430-51.
https://doi.org/10.1111/bph.15806 |
| 48 | Beamer CA, Seaver BP, Shepherd DM. Aryl hydrocarbon receptor (AhR) regulates silica-induced
inflammation but not fibrosis. Toxicol Sci. 2012;126(2):554-68.
https://doi.org/10.1093/toxsci/kfs024 |
| 49 | Amamou A, Yaker L, Leboutte M, Bôle-Feysot C, Savoye G, Marion-Letellier R. Dietary AhR
Ligands
Have No Anti-Fibrotic Properties in TGF-β1-Stimulated Human Colonic Fibroblasts. Nutrients.
2022;14(16). DOI: 10.3390/NU14163253
https://doi.org/10.3390/nu14163253 |
| 50 | Stockinger B, Meglio P Di, Gialitakis M, Duarte JH. The Aryl Hydrocarbon Receptor:
Multitasking
in
the Immune System. Annu Rev Immunol. 2014;32:403-432.
https://doi.org/10.1146/annurev-immunol-032713-120245 |
| 51 | Wajda A, Łapczuk J, Grabowska M, Pius-Sadowska E, Słojewski M, Laszczynska M, Urasinska E,
Machalinski B, Drozdzik M. Cell and region specificity of Aryl hydrocarbon Receptor (AhR) system
in
the testis and the epididymis. Reprod Toxicol. 2017;69:286-96.
https://doi.org/10.1016/j.reprotox.2017.03.009 |
| 52 | Al-Ghezi ZZ, Singh N, Mehrpouya-Bahrami P, Busbee PB, Nagarkatti M, Nagarkatti PS. AhR
Activation
by TCDD (2,3,7,8-Tetrachlorodibenzo-p-dioxin) Attenuates Pertussis Toxin-Induced Inflammatory
Responses by Differential Regulation of Tregs and Th17 Cells Through Specific Targeting by
microRNA.
Front Microbiol. 2019;10:2349.
https://doi.org/10.3389/fmicb.2019.02349 |
| 53 | Chang X, Fan Y, Karyala S, Schwemberger S, Tomlinson CR, Sartor MA, Puga A. Ligand-Independent
Regulation of Transforming Growth Factor β1 Expression and Cell Cycle Progression by the Aryl
Hydrocarbon Receptor. Mol Cell Biol. 2007;27(17):6127-39.
https://doi.org/10.1128/MCB.00323-07 |
| 54 | Yan J, Tung H-C, Li S, Niu Y, Garbacz WG, Lu P, Bi Y, Li Y, He J, Xu M, Ren S, Monga SP,
Schwabe
RF, Yang D, Xie W. Aryl Hydrocarbon Receptor Signaling Prevents Activation of Hepatic Stellate
Cells
and Liver Fibrogenesis in Mice. Gastroenterology. 2019;157(3):793-806.e14.
https://doi.org/10.1053/j.gastro.2019.05.066 |
| 55 | Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, MacDonald TT, Pallone F,
Monteleone
G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit
inflammation
in the gastrointestinal tract. Gastroenterology. 2011;141(1):237-48, 248.e1.
https://doi.org/10.1053/j.gastro.2011.04.007 |
| 56 | Lee C-C, Yang W-H, Li C-H, Cheng Y-W, Tsai C-H, Kang J-J. Ligand independent aryl hydrocarbon
receptor inhibits lung cancer cell invasion by degradation of Smad4. Cancer Lett.
2016;376(2):211-7.
https://doi.org/10.1016/j.canlet.2016.03.052 |
| 57 | Nakano N, Sakata N, Katsu Y, Nochise D, Sato E, Takahashi Y, Yamaguchi S, Haga Y, Ikeno S,
Motizuki M, Sano K, Yamasaki K, Miyazawa K, Itoh S. Dissociation of the AhR-ARNT complex by
TGF-β-Smad signaling represses CYP1A1 gene expression and inhibits benze[a]pyrene-mediated
cytotoxicity. J Biol Chem. 2020;295(27):9033-9051.
https://doi.org/10.1074/jbc.RA120.013596 |
| 58 | Kang JS, Kim HN, Jung DJ, Kim JE, Mun GH, Kim YS, Cho D, Shin DH, Hwang YI, Lee WJ. Regulation
of
UVB-Induced IL-8 and MCP-1 Production in Skin Keratinocytes by Increasing Vitamin C Uptake via
the
Redistribution of SVCT-1 from the Cytosol to the Membrane. J Invest Dermat. 2007;127(3):698-706.
https://doi.org/10.1038/sj.jid.5700572 |
| 59 | Lee WJ, Jo SY, Lee MH, Won CH, Lee MW, Choi JH, Chang SE. The Effect of MCP-1/CCR2 on the
Proliferation and Senescence of Epidermal Constituent Cells in Solar Lentigo. Int J Mol Sci.
2016;17(6):948.
https://doi.org/10.3390/ijms17060948 |
| 60 | Distler JHW, Akhmetshina A, Schett G, Distler O. Monocyte chemoattractant proteins in the
pathogenesis of systemic sclerosis. Rheumatology. 2009;48(2):98-103.
https://doi.org/10.1093/rheumatology/ken401 |
| 61 | Jiang WG, Sanders AJ, Ruge F, Harding KG. Influence of interleukin-8 (IL-8) and IL-8 receptors
on
the migration of human keratinocytes, the role of PLC-γ and potential clinical implications. Exp
Ther Med. 2012;3(2):231-6.
https://doi.org/10.3892/etm.2011.402 |
| 62 | Carvajal-Gonzalez JM, Roman AC, Cerezo-Guisado MI, Rico-Leo EM, Martin-Partido G,
Fernandez-Salguero PM. Loss of dioxin-receptor expression accelerates wound healing in vivo by a
mechanism involving TGFβ. J Cell Sci. 2009;122(11):1823-33.
https://doi.org/10.1242/jcs.047274 |
| 63 | Kuenzel NA, Dobner J, Reichert D, Rossi A, Boukamp P, Esser C. Vδ1 T Cells Integrated in
Full-Thickness Skin Equivalents: A Model for the Role of Human Skin-Resident γδT Cells. J Invest
Dermat. 2025;145(6):1407-21.
https://doi.org/10.1016/j.jid.2024.08.037 |