Original – DOI: 10.33594/000000855
Accepted 16.02.2026 - Published online 09.03.2026
Cellular Physiology and Biochemistry (60): 103 - 115
Regulation of Cellular Signaling by CUL5 is Dependent on Its Neddylation Status
Keywords
Abstract
Background/Aims: CUL5 acts as the scaffold protein of the E3 ligase complex in the ubiquitin-dependent protein degradation pathways. Overexpression of CUL5 inhibits cellular proliferation, whereas inhibition of CUL5 expression induces proliferation and can lead to the development of clinical disorders, including cancer. The effects of CUL5 depend on its post-translational modification by NEDD8 (neddylation), a process that represents a potential therapeutic target. This study explores the structure-function relationship between CUL5 and its neddylation status in vitro. Methods: CUL5 was mutated at the putative neddylation site Lys (K) 724 to Arg (K724RCUL5) and at three potential neddylation sites, K724, K727, and K728 (K724R/K727R/K728RCUL5). Because mutation of the PKA-phosphorylation site Ser (S) 730 (S730ACUL5) was previously shown to induce neddylation, a K724R/S730ACUL5 mutant was also generated. Mutant and wild type constructs were expressed in rat endothelial cells (RAMEC), T47D cancer cells, and COS-1 cells. Cellular proliferation, MAPK phosphorylation, ERα expression, and CUL5 neddylation status were analyzed, including treatment with the neddylation inhibitor MLN4924. Results: Expression of wild type (wt) CUL5 attenuated cell growth in RAMEC, T47D, and COS-1 cells. In contrast, expression of K724RCUL5 and K724R/S730ACUL5 mutants induced cellular growth, whereas the K724R/K727R/K728RCUL5 mutant had no significant effect on proliferation. In T47D cells, MAPK phosphorylation and estrogen receptor (ERα) expression were directly correlated with the neddylation status of CUL5. Western blot analysis of COS-1 cells treated with MLN4924 demonstrated that CUL5 remained neddylated in all mutant cell lines. Conclusion: These findings suggest that modification of CUL5 by NEDD8 may occur at multiple lysine residues and that multi-site neddylation may contribute to the diverse regulatory effects of CUL5 on cellular signaling and proliferation.
Introduction
Cullin 5 (CUL5), a 780 amino acid protein with a calculated Mr of 91 kDa, is the least conserved member of the cullin protein family, but it is highly homologous between species [1, 2]. CUL5 functions as a “core scaffold” involved in the formation of the E3-specific ligase complexes (Cullin-Ring Ligases - CRLs) to target proteins for Ubiquitin-Proteasome System (UPS) dependent degradation [3-8]. Human CUL5 localizes at chromosome 11 to the region at 11q22-23 [9] and the loss of heterozygosity on 11q22-23 CUL5 sequence has been linked to several distinct clinical disorders including cancers [10-15]. To characterize functions of CUL5, first identified as Vasopressin-Activated Calcium-Mobilizing protein VACM-1 [16], we demonstrated that over-expression of cul5 cDNA in several cell lines attenuated cellular proliferation by a mechanism that involves many processes: a decrease in adenylyl cyclase activity and cAMP production, a decrease in mitogen-activated protein kinase (MAPK) phosphorylation, a decrease in the nuclear early growth response element (egr-1) expression, and an increase in p53 and maspin proteins concentrations, pathways that regulate cellular growth in response to multiple pathophysiological stimuli [17-21]. In addition, we have reported that CUL5 has the ability to regulate estrogen-dependent growth in a T47D breast cancer derived cell line, as overexpression of CUL5 attenuated the expression of estrogen receptor (ERα) [22]. Further, human endothelial cells transfected with anti-cul5 siRNA had decreased endogenous CUL5 concentration and increased cellular growth when compared to the scrambled siRNA treated cells [23, 24]. Interestingly, others have shown that miRNA 19, which is upregulated in many cancers, targets CUL5 expression, further underscoring the importance of CUL5 in the regulation of cell function [25- 26]. CUL5 has also been implicated in the regulation of Src activity, cytokine signaling and viral infections [11, 27-30], and has been shown to be required for the degradation of several HSP90 clients that include oncogenic proteins activated in malignancies [31, 32]. It is now apparent that CUL5 activity is regulated by its posttranslational modification by NEDD8 (Neural precursor cell-Expressed Developmentally Down-regulated protein), an 81 amino acids residue protein originally isolated from mouse neural precursor cells, which is 80% homologous to ubiquitin [6, 7, 33, 34]. Neddylation involves a glycine residue located on the C-terminus of Nedd8 becoming covalently linked to a lysine residue on the protein substrate. Ligation of NEDD8 shifts the equilibrium to favor flexible open conformation of CRL that has a higher affinity for adaptor-bound substrates [29, 35-37]. While the complete mechanism behind neddylation has not yet been elucidated, studies suggest that a continuous and regulated cycle of neddylation and deneddylation is essential for the formation and activity of cullin-based ubiquitin ligases, which control ubiquitination of about 20% of cellular proteins destined for the degradation [36, 38]. In addition, the loss of NEDD8 function through drug inhibition or site-directed mutagenesis of the consensus neddylation site in cullins leads to changes in CRLs activities [29, 34, 39]. For example, CUL5 neddylation is required to target p53 for proteasomal degradation induced by adenoviral protein complexes in vitro, and increased neddylation has been associated with a poor prognosis of cancers [40-43]. Our previous studies suggested that many of the cellular functions of CUL5 are governed by its neddylation status, as the concentration of MAPK-P was decreased in the absence of neddylated CUL5, and the expression of the water channel aquaporin 1 (AQP1), which has been implicated in cancer development and tumor angiogenesis, in COS-1 cells was directly correlated to the neddylated CUL5 levels [17]. As recently reviewed by Zhang et al., [36], the neddylation pathway is tightly regulated in order to maintain protein homeostasis and proper control of broad cellular processes. Dysregulation of neddylation and de-neddylation pathways is associated with many human diseases including metabolic disorders, cardiac diseases, immune-related diseases and most importantly, cancer [ 36]. The most characterized substrate for the CUL5-specific CRL activity is APOBEC3G protein, which is essential in the prevention of HIV infectivity, where viral protein vif interacts with CUL5 based E3 ligase to induce APOBEC3G degradation [28, 34, 40]. Whereas the predicted neddylation site in CUL5 has been localized to K724 [43] studies examining the dependence of vif regulated degradation of APOBEC3G used a triple K mutant (K724RK727R/K728RCUL5) of CUL5 to generate a dominant negative phenotype that is not neddylated [28, 44]. Therefore, we hypothesized that, in addition to the consensus Lys (K) 724, other Lys residues in CUL5 can be neddylated and may alter CUL5-dependent control of specific signaling pathways. Such model is not unprecedented, as neddylation of pVHL, p53, parkin and E2F, occurs at numerous Lys residues, implying specificity and complexity of the NEDD8-dependent regulation of cell function [42, 36, 45-47]. Indeed, 1101 unique neddylation sites on 620 proteins have been identified [48], further suggesting the neddylation process regulates diverse biological signaling pathways. Whereas the inhibition of NEDD8-activating enzyme is already a promising new target for drug development against a broad spectrum of maladies that include cancer, cardiovascular, neurological and immune system pathologies [4, 40, 49-58], many questions regarding the mechanism and the control of the neddylation process remain unanswered. In this study, specific Lys (K) sites were mutated to Arg (R) to generate a construct where one lysine was mutated (K724RCUL5) and a construct where three lysine residues were mutated (K724R/K727R/K728RCUL5). Since we have shown previously that the mutation of the PKA-phosphorylation site in CUL5 induced its neddylation [23], we also generated a K724R/S730ACUL5 mutant. Our data suggest that mutation of the consensus neddylation site K724R does not completely prevent neddylation of CUL5 and does not inhibit its effect on cellular growth. Interestingly, our results suggest that K724R/K727ARK728RCUL5 mutant may still be neddylated, but it no longer affects cellular growth. Finally, protein analysis of the CUL5 sequence using NeddyPreddy program [59] revealed that, in addition to K724, K727 and K728 other K sites may be targets for neddylation. Thus, finding possible alternative neddylation sites in CUL5 and determining how they affect its localization and biological activity is important in identifying structure-based targets in CUL5 for the development of novel compounds to control cell functions and underlying clinical disorders.
Materials and Methods
Vectors/cDNA Constructs and Site-Directed Mutagenesis Site-directed mutagenesis in CUL5 sequence was performed using QuickChangeTM Kit from Stratagene (La Jolla, Ca) as previously reported [21]. Briefly, the mutagenesis primers were synthesized by IDT Inc. (Coralville, IA) and the mutation site sequences were confirmed by sequencing performed at the University of Michigan Core Facility. Wild type (wt) Cul5 and its mutants, S730ACUL5, K724RCUL5 K724R/S730ACUL5 or K724R/K727R/K728RCUL5 cDNAs were subcloned into the SalI/NotI restriction site in the polylinker region of pBK-CMV vector (Stratagene Co., La Jolla, Ca).
Antibodies We have previously demonstrated that polyclonal antibody A (Ab A) and Ab B, we generated against the amino and the carboxy termini of the CUL5 protein, respectively, immuno-precipitate the in vitro translated CUL5 protein, and that Cul5 cDNA transfected cells are immunostained with these antibodies while mock transfected controls remain unstained [16]. Both polyclonal and monoclonal anti-NEDD8 and anti-actin antibodies were purchased from either Abcam Co. or Sigma-Aldrich Co. MAPK (p44/42) and ERα antibodies were purchased from Cell Signaling Co. HRP conjugated secondary antibodies were purchased from Cell Signaling (Beverly, Ma). FITC and Texas Red conjugated secondary antibodies were purchased from Vector Laboratories (Burlingame, CA). LiCor antibodies were applied when the LiCor Odyssey Fc system was used for the Western blot signal detection.
Tissue Culture and Cellular Proliferation Assays Rat Adrenal Medulla Endothelial Cells (RAMEC) were cultured in DMEM low-glucose media supplemented with 10% Fetal Bovine Serum (FBS) and 1% Pen/Strep. T47D cells were cultured in RPMI 1640 media supplemented with 10% FBS, 0.1% Pen/Strep (10, 000 U/ml penicillin and 1000 mg/ml streptomycin) and 7.5 μg/ml insulin. Cells were plated at a density of 4-6x105 cells per 100 mm plate and maintained at 37°C under water-saturated 5% CO2. COS-1 cells were cultured in DMEM high-glucose media supplemented with 10% FBS and 1% Pen/Strep. The media was changed every 3-4 days. Cells transfected with the wt Cul5 cDNA and its mutants described above were selected with 50 μg/ml Geneticin as previously described [23]. Cell growth was monitored using the alarmarBlue® assay as described in the manufacturer’s instructions (Invitrogen Co.) and reported previously [24]. For the 96-well growth assay, cells were plated at 1.0x103 cells/well. After treatments, 10 μL of alamarBlue® reagent was added to each well and fluorescence readings at 595 nm were taken at 0, 24, 32, and 48 hours post-treatment using a Victor V3 plate reader (Perkin Elmer Co.).
Drug Treatment MLN4924, a NEDD8-activating enzyme inhibitor (Active Biochem Co), was used to inhibit neddylation of VACM-1/CUL5 [59]. Cells grown in 96-well plates (1.0x104 cells/ml) were treated with 20 µg/ml of MLN4924 and incubated at 37°C and 5% CO2 for the specified time periods.
In vitro Tumorigenicity Assays Matrigel® (Becton Dickinson Co,) invasion assays were performed in 96 well plates coated with GFR (Growth Factor Reduced) Matrigel® support at a medium thickness described in the manufacturer's instructions. Cells were plated at 5x104 and 5x105 cells per well and incubated at 37°C with 5% CO2 for the specified time period. For quantitation, randomly selected fields were photographed by phase contrast microscopy. Each experiment was repeated at least three times.
Immunostaining Indirect immunofluorescence for CUL5 and NEDD8 was used to stain cells grown on glass coverslips. Cells were plated into 6 or 12-well plates on coverslips at a density of 1x104 cells per well and grown for 3-5 days. Cells were fixed in 3% paraformaldehyde (in phosphate buffered saline, PBS, pH 7.4) for 20 minutes, washed in 3% BSA made in PBS, permeabilized with 0.5% Tween-20 (in 1X PBS) for 20 minutes, washed with 3% BSA/PBS, and incubated for one hour with primary anti-CUL5, anti-ERα or NEDD8 specific antibodies diluted in PBS with 3% BSA. Coverslips were then washed with PBS/3% BSA/PBS solution and incubated with secondary antibodies (in 1X PBS/3% BSA) for one hour. Fluorescein isothiocyanate-conjugated anti-rabbit and Texas Red-conjugated anti-mouse Ab (Vector Laboratories Inc., Burlingame, CA) were used as appropriate. Phalloidin stain (Sigma Co.) was used to show actin expression. The coverslips were washed again with 3% BSA/PBS and mounted onto slides using Vectashield mounting medium with DAPI (Vector Laboratories Inc.). The slides were imaged using epifluorescence microscopy (Nikon Instruments Incl.; Melville, NY).
Western Blotting RAMEC, T47D and Cos-1 cells grown to appropriate confluency were washed with and resuspended in ice-cold PBS. Cells were centrifuged at 1500 RPM for 5 minutes, and the pellet was resuspended in 250 μl of Morgan buffer (5ml of 1M Tris, 100 μl Triton-X-100, 5 ml of 3 M NaCl, 200 μl of 500 mM EDTA, 10 mls of 800 mM NaF, and ddH2O to 100 ml. The suspension was homogenized with a Polytron homogenizer and protein concentration in all samples was determined using either a Cary50 or Shimadzu spectrophotometers. Total cell lysates were resuspended in 4X sample buffer (Invitrogen Co), heated to 70°C for 5 min, and subjected to SDS polyacrylamide gel electrophoresis using a Novex 4-12% Bis-Tris gel (Invitrogen Co). The separated proteins were transferred onto nitrocellulose membranes (Thermo Scientific Co.) by wet transfer at a constant 30 V for 2 hrs. Non-specific binding was blocked with 5% non-fat dry milk in PBS for 30 min on a shaker at room temperature. All washes were done in ECL solution (4g NaCl, 10 mls of 1M Tris, 250 µl Tween-20 and ddH2O to 500 ml). The membranes were incubated with primary antibodies described above on a shaker for 1.5 hrs at room temperature and subsequently with Li-COR secondary antibodies for 1 hr at room temperature and the visualization of bands was carried out with the Li-COR Fc scanner. To correct for equal protein loading, blots were stripped and re-probed with anti-actin or GAPDH antibodies (Abcam, Cambridge, MA). All immunoblots and Western blots were scanned, and the signal was quantitated using the NIH Imaging Analysis Program (http://rsb.info.nih.gov/ij/index.html).
Statistics Data are expressed as means ± one standard error (SE) of the mean. The ANOVA and Student's t-test were used for statistical analysis with significance set at p < 0.05.
Results
Cullin sequence analysis using NCBI data analysis program (BLAST) identified the presence of the putative neddylation site in all cullins (Fig 1A) [7]. In CUL5, the predicted neddylation site was found to localize to Lys 724 in several species examined (Fig. 1B). To determine the effects of neddylation on CUL5 activity, we mutated the consensus neddylation site, Lys724 to Arg in wt Cul5 (K724RCUL5). Since we have shown previously that mutation of the PKA-phosphorylation site Ser (S) 730 (S730ACUL5) in CUL5 induced its neddylation [23], we also generated a K724R/S730ACUL5 mutant (Fig. 1C-D). Expression of wild type (wt) CUL5 in rat endothelial cells (RAMEC) attenuated cell growth, whereas expression of mutant constructs induced cellular proliferation (Fig. 1E). Phalloidin staining demonstrated altered actin organization and growth patterns in cells transfected with mutated Cul5 cDNA compared to CMV controls (Fig. 1F). In T47D cancer cells, expression of wt CUL5 significantly decreased cellular proliferation (Fig. 2A). In contrast, expression of S730ACUL5, K724R/S730ACUL5, and K724RCUL5 significantly induced proliferation compared to control cells (Fig. 2A). No apparent morphological differences were observed when these cells were grown on polystyrene dishes (Fig. 2B). We next examined whether mutation of neddylation sites affected regulation of MAPK and ERα signaling pathways. The level of MAPK phosphorylation was directly proportional to the ratio of NEDD8CUL5/CUL5 (R² = 0.60) (Figs. 2C and 2D). Similarly, ERα levels were directly correlated with CUL5 neddylation status (R² = 0.62) (Fig. 2F). When ERα levels were plotted against the ratio of NEDD8CUL5/CUL5, the correlation coefficient increased further (R² = 0.85) (Fig. 2G). To further investigate neddylation, we generated a triple lysine mutant, K724R/K727R/K728RCUL5 (3K) (Fig. 3A-B), previously used in studies of vif-regulated degradation of APOBEC3G [40]. In RAMEC cells, expression of the 3K mutant no longer affected cell growth compared to wt CUL5 (Fig. 3C). Western blot analysis revealed persistent NEDD8 signal in K724R/K727R/K728RCUL5-expressing cells (Fig. 3D). When cells were plated on Matrigel® support, wt CUL5 expression attenuated cord formation, whereas both K724RCUL5 and the 3K mutant enhanced cord formation compared to controls (Fig. 3E). This effect depended on both cell density and duration of culture on Matrigel® (Fig. 3E). To validate these findings, we expressed a K724A/K727A/K728ACUL5 (3K) mutant in COS-1 cells. Cells were plated at equal densities (7×10⁵ cells/plate) and counted after three days. Immunocytochemistry confirmed expression of both Cul5 and NEDD8 in all cell lines (Fig. 4A). As observed in RAMEC, wt CUL5 reduced proliferation in COS-1 cells, whereas the 3K mutant did not affect proliferation compared to CMV controls (Fig. 4B). Wound-healing assays confirmed reduced re-growth in wt CUL5-expressing cells but not in 3K mutant cells (Fig. 4C). Western blot analysis demonstrated persistent NEDD8-CUL5 signal in 3K mutant cells (Fig. 4D). Treatment with MLN4924 (0.33 µM), an inhibitor of the NEDD8 E1 activating enzyme [38, 59], reduced NEDD8 signal intensity in mutant cells (Fig. 4E), confirming that the detected band represented neddylated CUL5. Similar effects were observed in other mutant cell lines (Fig. 4F-G). Finally, the NeddyPreddy program [58] predicted several additional putative neddylation sites in CUL5 with strong probability scores (Fig. 5A-B).
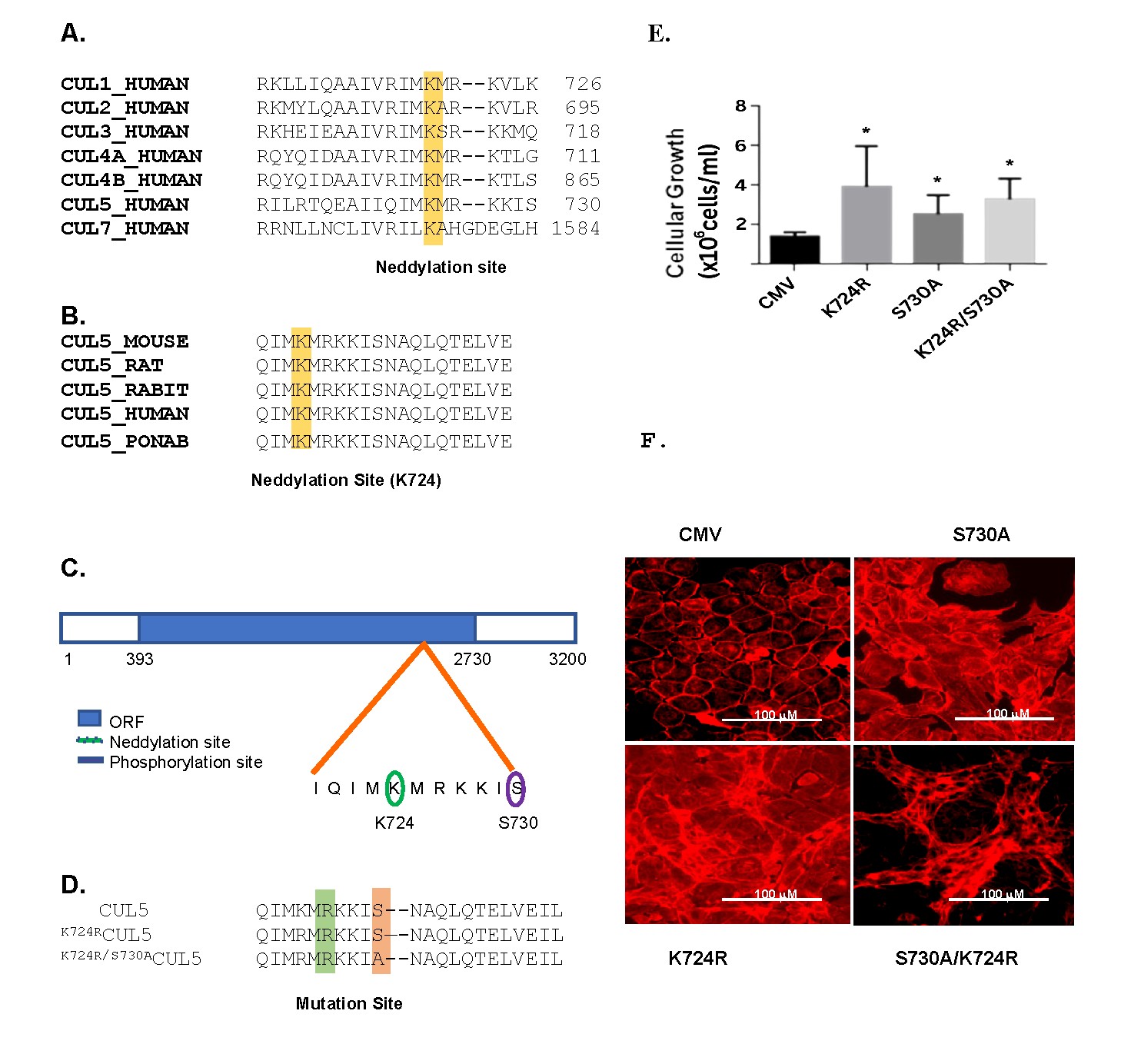
Fig. 1: Fig. 1. Identification and alignment of the putative neddylation site (K724) in cullins. (A) Alignment of the amino acid sequences of human cullins using NCBI databases analysis. The putative neddylation Lys 724 is highlighted in yellow. The position of the last amino acid shown is marked on the right. (B) Alignment of CUL5 putative neddylation sequence in five eukaryotic organisms (mouse, rat, rabbit, human, and orangutan (Pongo abelii), where the putative consensus neddylation site localizes to K724, and is highlighted in yellow. (C) Schematic diagram of CUL5 open reading frame (ORF, NT: 393 to 2730, solid blue) and approximate locations of the consensus sequences for a NEDD8 modification at K724 (green oval) and a PKA-dependent phosphorylation site at S730 (purple oval). (D) The alignment of the wt CUL5 and CUL5 where Lys 724 was mutated to Arg (K724RCUL5) (highlighted green) and CUL5 where Lys724 and Ser730 (highlighted orange) were mutated to Arg and Ala, respectively (K724R/S730ACUL5). (E) Cellular growth rate in RAMEC transfected with either the CMV vector, the CMV vector containing cul5 or mutant cul5 cDNAs (K724RCUL5, S730ACUL5, and K724R/S730ACUL5) as described in the Methods. (n=4 per group, *, p<0.05 when compared to the CMV). (F) Cell lines stained with phalloidin reagent which is specific for actin, showed phenotypic change in growth pattern between cell lines described above.
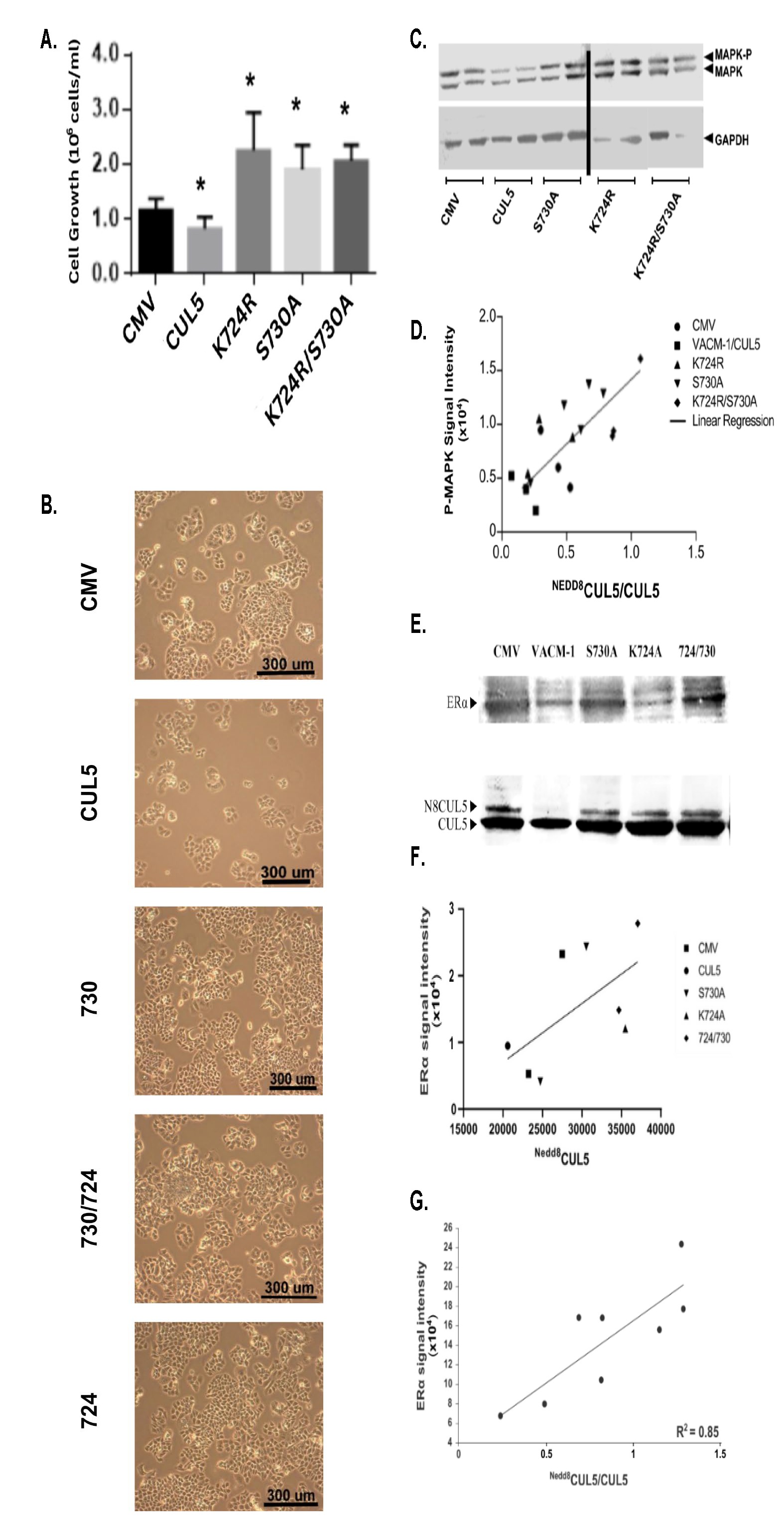
Fig. 2: Fig. 2. Effects of CUL5 and its mutants on cellular proliferation, phosphorylation of MAPK (MAPK-P) and estrogen receptor (ERα) levels in T47D cancer cells. (A) T47D cells were transfected with a vector (CMV), CUL5, K724RCUL5, S730ACUL5 and K724R/S730ACUL5 cDNA mutants and cellular growth was determined using alamarBlue® assay, as described in Methods (n=4 per group, * p<0.05 from CMV). (B) Cellular proliferation of the T47D cell lines transfected with the constructs described above, plated on polystyrene support and monitored using a phase contrast microscope (Magnification, 20X). (C) Representative Western blot of MAPK and phosphorylated MAPK (p44/p42) (MAPK-P) protein levels in lysates from cells transfected with the wt and mutated CUL5. To correct for unequal loading, blots were stripped and re-probed with an anti-GAPDH antibody (lower bands) (n=3 per group). (D) MAPK-P signal intensities from C were quantified and plotted against the ratio of NEDD8CUL5/CUL5 signal (R2=0.60) (E) Representative Western blot analysis of lysates from T47D cell lines probed with anti-ERα antibody and reprobed with anti CUL5 antibody (below). (F) ERα signal intensity observed on Western blot analysis was plotted against the NEDD8CUL5 signal. (n=3, R2=0.62). (G) ERα signal intensity observed on Western blot analysis plotted against the ratio of NEDD8CUL5/CUL5 signal (R2 =0.85).
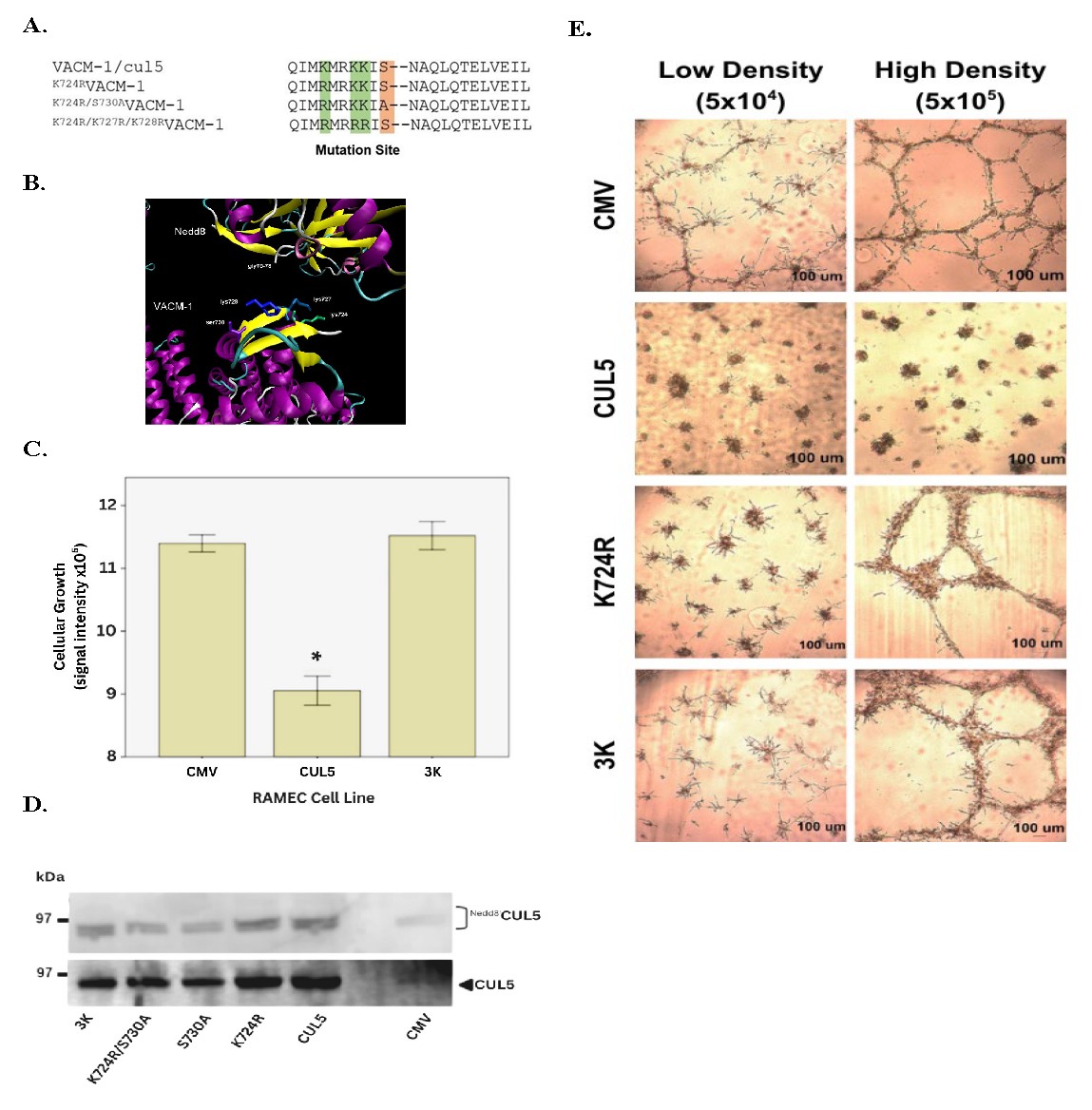
Fig. 3: Fig. 3. Effects of K724R/K727R/K728RCUL5 (3K) mutant on cellular proliferation in RAMEC. (A) Alignment of CUL5, K724RCUL5, K724R/S730ACUL5 and K724R/K727R/K728RCUL5 (3K) mutants. (B) CUL5-model identifying potential neddylation sites generated using PyMol program. (C) Cellular proliferation of RAMEC transfected with CMV vector, CUL5 cDNA or K724R/K727R/K728RCUL5 (3K) (n= 6 per group, *, p<0.05 when compared to CMV. (D) Western blot analysis of lysates prepared from mutant-transfected RAMEC and probed with anti-NEDD8 (upper band) and anti-CUL5 (lower band) antibodies as described in Methods. (E) Growth pattern of RAMEC transfected with CMV vector, CUL5, K724RCUL5, K724R/K727R/K728RCUL5 and plated at a different density on a Matrigel® support.
Fig. 4: Fig. 4. Effects of K724R/K727R/K728RCUL5 (3K) mutant on CUL5 neddylation and on cellular proliferation in COS-1 cells. A. Immunocytochemistry of COS-1 cells transfected with CUL5 and 3K mutant cDNAs demonstrate the expression of CUL5 and NEDD8 in all three cell lines. (B) Cellular growth between CMV, wt-CUL5 and K724R/727R/K728RCUL5 (3K) cDNA mutant was compared at 24 hrs (n=3 per group, * p<0.05). (C) Cellular proliferation of cos 1 cells transfected with vector (CMV), wt-CUL5, K724RCUL5, S730ACUL5, and K724R/S730ACUL5 cDNA mutants was determined using a “wound assay” approach (n=3 per group, * p<0.05). (D) Representative Western blot analysis of CUL5 expression in CMV, CUL5 and K724R/727R/K728RCUL5 (3K) transfected COS-1 cells. (E) Effects of MLN4924 on the neddylation status of CUL5 in K724R/K727R/K728RCUL5 (3K) cells (n=2). (F) Effects of MLN4924 on the CUL5 neddylation status in vector (CMV), wt-CUL5, S730ACUL5, K724R/S730ACUL5 cDNA and K724R/K727R/K728RCUL5 (3K) transfected COS-1 cells. (G) Data shown in F were corrected for actin levels and expressed as a ratio.
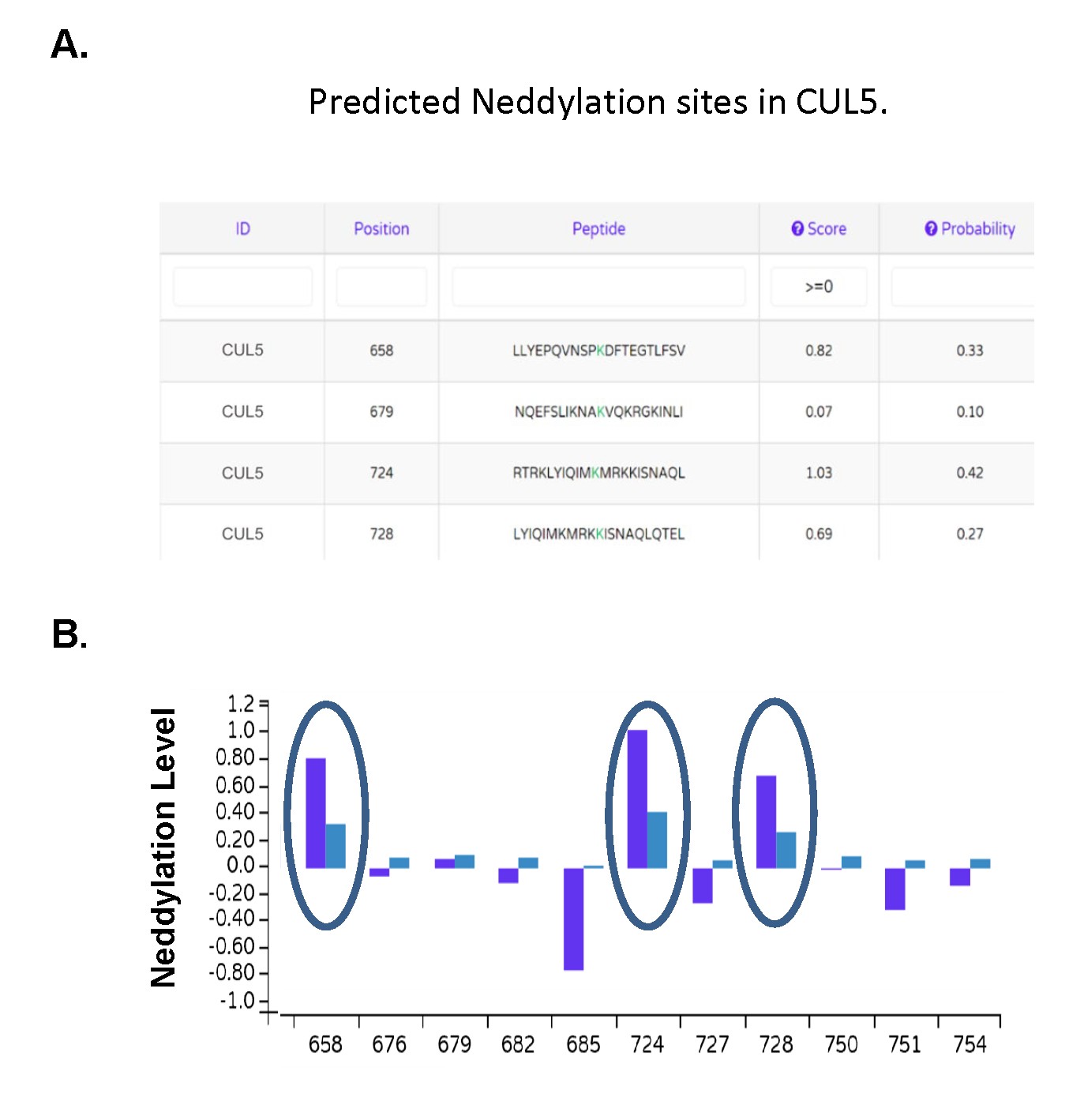
Fig. 5: Fig. 5. Identification of potential neddylation sites in CUL5. (A) The NeddyPreddy program was used to identify potential new neddylation sites in CUL5 (59). (B) Dark blue bars indicate the neddylation score and light blue bars the probability levels.
Discussion
Our findings indicate that although the predicted consensus neddylation site in CUL5 is Lys724 [7], mutation of this site does not abolish CUL5 neddylation. Even mutation of three lysine residues (K724/K727/K728) did not eliminate the NEDD8-modified band, suggesting that CUL5 may undergo multi-site neddylation. The observed correlation between CUL5 neddylation status and MAPK phosphorylation or ERα expression suggests a regulatory relationship between neddylation and downstream signaling. However, these data demonstrate correlation rather than direct mechanistic causation. The triple mutant lost the growth-suppressive function observed with wt CUL5, despite remaining neddylated. This finding suggests that either alternative lysine residues are modified or that specific lysine residues may differentially regulate CUL5 biological activity. The persistence of NEDD8 signal following lysine mutations and its reduction after MLN4924 treatment [38, 59] further supports the conclusion that CUL5 can be modified at multiple lysine residues. This concept is consistent with reports that other proteins, including E2F1 and p53, undergo multi-site neddylation [40]. Bioinformatic prediction using NeddyPreddy [58] further supports the possibility of additional modification sites in CUL5. Collectively, these findings suggest a more complex model of CUL5 regulation than previously appreciated. Nevertheless, the present study does not directly identify the alternative lysine residues involved. Additional studies, including site mapping and proteomic analyses, will be necessary to define site-specific modification patterns and their functional consequences. In conclusion, while cullins are generally reported to contain a single neddylation site (Fig. 1), our data suggest that CUL5 may be neddylated at multiple lysine residues, which may contribute to its diverse regulatory effects on cellular signaling and proliferation.
Conclusion
Although cullins are reported to have only one neddylation site (Fig 1), our work suggests that CUL5 may have more than one neddylation site involved in the control of its biological activity. These results are not unprecedented as numerous other proteins are neddylated at multiple Lys sites. For example, E2F1 has 13 neddylation sites, and multi-site neddylation is required to target p53 for proteasomal degradation induced by adenoviral protein complexes in vitro [40]. Clearly, further studies are required to determine how site-specific neddylation(s) of CUL5 affect its function that leads to the broad effect on cellular processes.
Acknowledgements
This work was supported by the Schaap Endowed Funds for undergraduate research and by Frederich Garrett and Helen Floor Dekker Endowed Award. All authors except for MBH were undergraduate students at Hope College when performing this work. We also acknowledge the editorial assistance of Dr. Chris Barney and Elizabeth Hinkley.
Disclosure Statement
The authors have nothing to disclose.
References
- Kipreos, E. T., Lander, L. E., Wing, J. P., He, W. W., & Hedgecock, E. M. (1996). Cul-1 is required for cell cycle exit in C. elegans and identifies a novel gene family. Cell, 85(6), 829–839. https://doi.org/10.1016/s0092-8674(00)81267-2
- Mathias, N., Johnson, S. L., Winey, M., Adams, A. E. M., Goetsch, L., Pringle, J. R., Byers, B., & Goebl, M. G. (1996). Cdc53p Acts in Concert with Cdc4p and Cdc34p To Control the G1-to-S-Phase Transition and Identifies a Conserved Family of Proteins. Molecular and Cellular Biology, 16(12), 6634–6643. https://doi.org/10.1128/MCB.16.12.6634
- Jones, J., Wu, K., Yang, Y., Guerrero, C., Nillegoda, N., Pan, Z.-Q., & Huang, L. (2008). A Targeted Proteomic Analysis of the Ubiquitin-Like Modifier Nedd8 and Associated Proteins. Journal of Proteome Research, 7(3), 1274–1287. https://doi.org/10.1021/pr700749v
- Petroski, M. D., & Deshaies, R. J. (2005). Function and regulation of cullin–RING ubiquitin ligases. Nature Reviews Molecular Cell Biology, 6(1), Article 1. https://doi.org/10.1038/nrm1547
- Duda, D. M., Borg, L. A., Scott, D. C., Hunt, H. W., Hammel, M., & Schulman, B. A. (2008). Structural insights into NEDD8 activation of cullin-RING ligases: Conformational control of conjugation. Cell, 134(6), 995–1006. https://doi.org/10.1016/j.cell.2008.07.022
- Hori, T., Osaka, F., Chiba, T., Miyamoto, C., Okabayashi, K., Shimbara, N., Kato, S., & Tanaka, K. (1999). Covalent modification of all members of human cullin family proteins by NEDD8 Oncogene, 18(48), Article 48. https://doi.org/10.1038/sj.onc.1203093
- Kamitani, T., Kito, K., Nguyen, H. P., & Yeh, E. T. (1997). Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. The Journal of Biological Chemistry, 272(45), 28557–28562. https://doi.org/10.1074/jbc.272.45.28557
- Bano, I., Soomro, A. S., Abbas, S. Q., Ahmadi, A., Hassan, S. S. ul, Behl, T., & Bungau, S. (2022). A Comprehensive Review of Biological Roles and Interactions of Cullin-5 Protein. ACS Omega, 7(7), 5615–5624. https://doi.org/10.1021/acsomega.1c06890
- Byrd, P. J., Stankovic, T., McConville, C. M., Smith, A. D., Cooper, P. R., & Taylor, A. M. (1997). Identification and analysis of expression of human VACM-1, a cullin gene family member located on chromosome 11q22-23 Genome Research, 7(1), 71–75. https://doi.org/10.1101/gr.7.1.71
- Fay, M. J., Longo, K. A., Karathanasis, G. A., Shope, D. M., Mandernach, C. J., Leong, J. R., Hicks, A., Pherson, K., & Husain, A. (2003). Analysis of CUL-5 expression in breast epithelial cells, breast cancer cell lines, normal tissues and tumor tissues. Molecular Cancer, 2(1), 40. https://doi.org/10.1186/1476-4598-2-40
- Laszlo, G. S., & Cooper, J. A. (2009). Restriction of Src activity by Cullin-5 Current Biology: CB, 19(2), 157–162. https://doi.org/10.1016/j.cub.2008.12.007
- Ceremuga, T. E., Yao, X.-L., Alam, H. B., & McCabe, J. T. (2003). Alterations of Cullin-5 mRNA levels in the rat central nervous system following hemorrhagic shock. Neurological Research, 25(2), 211–216. https://doi.org/10.1179/016164103101201229
- Ying, J., Zhang, M., Qiu, X., & Lu, Y. (2018). Targeting the neddylation pathway in cells as potential therapeutic approach for diseases. Cancer Chemotherapy and Pharmacology, 81(5), 797–808. https://doi.org/10.1007/s00280-018-3541-8
- Zhu, Z., Sun, L., Hao, R., Jiang, H., Qian, F., & Ye, R. D. (2017). Nedd8 modification of Cullin-5 regulates lipopolysaccharide-induced acute lung injury. American Journal of Physiology-Lung Cellular and Molecular Physiology, 313(1), L104–L114. https://doi.org/10.1152/ajplung.00410.2016
- Zou, T., & Zhang, J. (2021). Diverse and pivotal roles of neddylation in metabolism and immunity. The FEBS Journal, 288(13), 3884–3912. https://doi.org/10.1111/febs.15584
- Burnatowska-Hledin, M. A., Spielman, W. S., Smith, W. L., Shi, P., Meyer, J. M., & Dewitt, D. L. (1995). Expression cloning of an AVP-activated, calcium-mobilizing receptor from rabbit kidney medulla. The American Journal of Physiology, 268(6 Pt 2), 1198–1210. https://doi.org/10.1152/ajprenal.1995.268.6.F1198
- Van Dort, C., Zhao, P., Parmelee, K., Capps, B., Poel, A., Listenberger, L., Kossoris, J., Wasilevich, B., Murrey, D., Clare, P., & Burnatowska-Hledin, M. (2003). VACM-1, a cul-5 gene, inhibits cellular growth by a mechanism that involves MAPK and p53 signaling pathways. American Journal of Physiology-Cell Physiology, 285(6), C1386–C1396. https://doi.org/10.1152/ajpcell.00338.2002
- Willis, A. N., Dean, S. E. B., Habbouche, J. A., Kempers, B. T., Ludwig, M. L., Sayfie, A. D., Lewis, S. P., Harrier, S., DeBruine, Z. J., Garrett, R., & Burnatowska-Hledin, M. A. (2017). Nuclear localization signal sequence is required for VACM-1/CUL5-dependent regulation of cellular growth. Cell and Tissue Research, 368(1), 105–114. https://doi.org/10.1007/s00441-016-2522-7
- Burnatowska-hledin, M., Zeneberg, A., Roulo, A., Grobe, J., Zhao, P., Lelkes, P. I., Clare, P., & Barney, C. (2001). Expression of VACM-1 Protein in Cultured Rat Adrenal Endothelial Cells is Linked to the Cell Cycle. Endothelium, 8(1), 49–63. https://doi.org/10.3109/10623320109063157
- Burnatowska-Hledin, M. A., & Barney, C. C. (2014). New Insights into the Mechanism for VACM-1/cul5 Expression in Vascular Tissue in vivo. In K. W. Jeon (Ed.), International Review of Cell and Molecular Biology (Vol. 313, pp. 79–101). Academic Press. https://doi.org/10.1016/B978-0-12-800177-6.00003-7
- Buchwalter, A., Van Dort, C., Schultz, S., Smith, R., Le, I. P., Abbott, J. L., Oosterhouse, E., Johnson, A. E., Hansen-Smith, F., & Burnatowska-Hledin, M. (2008). Expression of VACM-1/cul5 mutant in endothelial cells induces MAPK phosphorylation and maspin degradation and converts cells to the angiogenic phenotype. Microvascular Research, 75(2), 155–168. https://doi.org/10.1016/j.mvr.2007.08.004
- Johnson, A. E., Le, I. P., Andresen, B. T., Stodola, J., Dewey, G. L., Dean, S. B., Resau, J., Haak, P., Ruch, T., Sartor, A., Lazdins, I., Barney, C. C., & Burnatowska-Hledin, M. A. (2012). VACM-1/cul5 expression in vascular tissue in vivo is induced by water deprivation and its expression in vitro regulates aquaporin-1 concentrations. Cell and Tissue Research, 349(2), 527–539. https://doi.org/10.1007/s00441-012-1419-3
- Bradley, S. E., Johnson, A. E., Le, I. P., Oosterhouse, E., Hledin, M. P., Marquez, G. A., & Burnatowska-Hledin, M. (2010). Phosphorylation of VACM-1/Cul5 by Protein Kinase A Regulates Its Neddylation and Antiproliferative Effect. The Journal of Biological Chemistry, 285(7), 4883–4895. https://doi.org/10.1074/jbc.M109.085225
- Kunkler, B., Salamango, D., DeBruine, Z. J., Ploch, C., Dean, S., Grossens, D., Hledin, M. P., Marquez, G. A., Madden, J., Schnell, A., Short, M., & Burnatowska-Hledin, M. A. (2018). CUL5 is required for thalidomide-dependent inhibition of cellular proliferation. PLOS ONE, 13(5), e0196760 https://doi.org/10.1371/journal.pone.0196760
- Xu, X.-M., Wang, X.-B., Chen, M.-M., Liu, T., Li, Y.-X., Jia, W.-H., Liu, M., Li, X., & Tang, H. (2012). MicroRNA-19a and -19b regulate cervical carcinoma cell proliferation and invasion by targeting CUL5. Cancer Letters, 2(322), 148–158. https://doi.org/10.1016/j.canlet.2012.02.038
- Liu, X. S., Chopp, M., Wang, X. L., Zhang, L., Hozeska-Solgot, A., Tang, T., Kassis, H., Zhang, R. L., Chen, C., Xu, J., & Zhang, Z. G. (2013). MicroRNA-17-92 cluster mediates the proliferation and survival of neural progenitor cells after stroke. The Journal of Biological Chemistry, 288(18), 12478–12488. https://doi.org/10.1074/jbc.M112.449025
- Wang, K., & Liu, X. (2022). Determining the Effects of Neddylation on Cullin-RING Ligase–Dependent Protein Ubiquitination. Current Protocols, 2(3), e401. https://doi.org/10.1002/cpz1.401
- Liu, B., Sarkis, P. T. N., Luo, K., Yu, Y., & Yu, X.-F. (2005). Regulation of Apobec3F and Human Immunodeficiency Virus Type 1 Vif by Vif-Cul5-ElonB/C E3 Ubiquitin Ligase. Journal of Virology, 79(15), 9579–9587 https://doi.org/10.1128/jvi.79.15.9579-9587.2005
- Wolfe, L. S., Stanley, B. J., Liu, C., Eliason, W. K., & Xiong, Y. (2010). Dissection of the HIV Vif Interaction with Human E3 Ubiquitin Ligase. Journal of Virology, 84(14), 7135–7139. https://doi.org/10.1128/jvi.00031-10
- Wormald, S., & Hilton, D. J. (2004). Inhibitors of cytokine signal transduction. The Journal of Biological Chemistry, 279(2), 821–824. https://doi.org/10.1074/jbc.R300030200
- Samant, R. S., Clarke, P. A., & Workman, P. (2014). E3 ubiquitin ligase Cullin-5 modulates multiple molecular and cellular responses to heat shock protein 90 inhibition in human cancer cells. Proceedings of the National Academy of Sciences, 111(18), 6834–6839. https://doi.org/10.1073/pnas.1322412111
- Ehrlich, E. S., Wang, T., Luo, K., Xiao, Z., Niewiadomska, A. M., Martinez, T., Xu, W., Neckers, L., & Yu, X.-F. (2009). Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase. Proceedings of the National Academy of Sciences, 106(48), 20330–20335. https://doi.org/10.1073/pnas.0810571106
- Saha, A., & Deshaies, R. J. (2008). Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Molecular Cell, 32(1), 21–31. https://doi.org/10.1016/j.molcel.2008.08.021
- Rabut, G., & Peter, M. (2008). Function and regulation of protein neddylation. EMBO Reports, 9(10), 969–976. https://doi.org/10.1038/embor.2008.183
- Deshaies, R. J., Emberley, E. D., & Saha, A. (2010). Control of Cullin-Ring Ubiquitin Ligase Activity by Nedd8. In M. Groettrup (Ed.), Conjugation and Deconjugation of Ubiquitin Family Modifiers: Subcellular Biochemistry (pp.41–56).Springer. https://doi.org/10.1007/978-1-4419-6676-6_4
- Zhang, S., Yu, Q., Li, Z., Zhao, Y., & Sun, Y. (2024). Protein neddylation and its role in health and diseases. Signal Transduction and Targeted Therapy, 9(1), 85. https://doi.org/10.1038/s41392-024-01800-9
- Lumpkin, R. J., Ahmad, A. S., Blake, R., Condon, C. J., & Komives, E. A. (2021). The Mechanism of NEDD8 Activation of CUL5 Ubiquitin E3 Ligases. Molecular & Cellular Proteomics: MCP, 20, 100019. https://doi.org/10.1074/mcp.RA120.002414
- Soucy, T. A., Smith, P. G., Milhollen, M. A., Berger, A. J., Gavin, J. M., Adhikari, S., Brownell, J.E., Burke, K. E., Cardin, D. P., Critchley, S., Cullis, C. A., Doucette, A., Garnsey, J. J., Gaulin, J. L., Gershman, R. E., Lublinsky, A. R., McDonald, A., Mizutani, H., Narayanan, U., Langston, S. P. (2009). An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature, 458(7239), Article 7239. https://doi.org/10.1038/nature07884
- Wu, J.-T., Lin, H.-C., Hu, Y.-C., & Chien, C.-T. (2005). Neddylation and deneddylation regulate Cul1 and Cul3 protein accumulation. Nature Cell Biology, 7(10), Article 10. https://doi.org/10.1038/ncb1301
- Yu, X., Yu, Y., Liu, B., Luo, K., Kong, W., Mao, P., & Yu, X.-F. (2003). Induction of APOBEC3G Ubiquitination and Degradation by an HIV-1 Vif-Cul5-SCF Complex. Science, 302(5647), 1056–1060. https://doi.org/10.1126/science.1089591
- Abidi, N., & Xirodimas, D. P. (2015). Regulation of cancer-related pathways by protein NEDDylation and strategies for the use of NEDD8 inhibitors in the clinic. Endocrine-Related Cancer, 22(1), T55–T70. https://doi.org/10.1530/ERC-14-0315
- Aoki, I., Higuchi, M., & Gotoh, Y. (2013). NEDDylation controls the target specificity of E2F1 and apoptosis induction. Oncogene, 32(34), Article 34. https://doi.org/10.1038/onc.2012.428
- Stastna, M., & Van Eyk, J. E. (2015). Posttranslational modifications of lysine and evolving role in heart pathologies—Recent developments. PROTEOMICS, 15(5–6), 1164–1180. https://doi.org/10.1002/pmic.201400312
- Stanley, D. J., Bartholomeeusen, K., Crosby, D. C., Kim, D. Y., Kwon, E., Yen, L., Cartozo, N. C., Li, M., Jäger, S., Mason-Herr, J., Hayashi, F., Yokoyama, S., Krogan, N. J., Harris, R. S., Peterlin, B. M., & Gross, J. D. (2012). Inhibition of a NEDD8 Cascade Restores Restriction of HIV by APOBEC3G. PLOS Pathogens, 8(12), e1003085 https://doi.org/10.1371/journal.ppat.1003085
- Feng, L., Lin, T., Uranishi, H., Gu, W., & Xu, Y. (2005). Functional Analysis of the Roles of Posttranslational Modifications at the p53 C Terminus in Regulating p53 Stability and Activity. Molecular and Cellular Biology, 25(13), 5389–5395. https://doi.org/10.1128/MCB.25.13.5389-5395.2005
- Querido, E., Blanchette, P., Yan, Q., Kamura, T., Morrison, M., Boivin, D., Kaelin, W. G., Conaway, R. C., Conaway, J. W., & Branton, P. E. (2001). Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes & Development, 15(23), 3104–3117. https://doi.org/10.1101/gad.926401
- Okumura, F., Joo-Okumura, A., Nakatsukasa, K., & Kamura, T. (2016). The role of cullin 5-containing ubiquitin ligases. Cell Division, 11(1), 1. https://doi.org/10.1186/s13008-016-0016-3
- Labata-Gil, S., Heidelberger, J.B., Maghames, C., Rodriguez, M.S., Beli, P., Xirodimas, D.P. (2021). Proteome-wide identification of NEDD8 modification sites reveals distinct proteomes for canonical and atypical NEDDylation. Cell Reports 34, 108635. https://www.cell.com/cell-reports/pdf/S2211-1247(20)31624-7.pdf
- Pan, Z.-Q., Kentsis, A., Dias, D. C., Yamoah, K., & Wu, K. (2004). Nedd8 on cullin: Building an expressway to protein destruction. Oncogene,: 23(11), Article 11. https://doi.org/10.1038/sj.onc.1207414
- Gao, F., Fan, Y., Zhou, B., Guo, W., Jiang, X., Shi, J., & Ren, C. (2020). The functions and properties of cullin-5, a potential therapeutic target for cancers. American Journal of Translational Research, 12(2), 618–632.
- Diaz, S., Wang, K., Sjögren, B., & Liu, X. (2022). Roles of Cullin-RING Ubiquitin Ligases in Cardiovascular Diseases. Biomolecules, 12(3), 416. https://doi.org/10.3390/biom12030416
- Li, J., Zou, J., Littlejohn, R., Liu, J., & Su, H. (2020). Neddylation, an Emerging Mechanism Regulating Cardiac Development and Function. Frontiers in Physiology, 11. https://www.frontiersin.org/articles/10.3389/fphys.2020.612927
- Li, H., Zhou, W., Li, L., Wu, J., Liu, X., Zhao, L., Jia, L., & Sun, Y. (2017). Inhibition of Neddylation Modification Sensitizes Pancreatic Cancer Cells to Gemcitabine. Neoplasia, 19(6), 509–518. https://doi.org/10.1016/j.neo.2017.04.003
- Li, Z., Hu, N., Dai, L., Hou, X., Hu, W., Liang, W., & Wang, X. (2021). Cullin-5 (CUL5) as a potential prognostic marker in a pan-cancer analysis of human tumors. Bioengineered, 12(1), 5348–5360. https://doi.org/10.1080/21655979.2021.1940042
- Vogl, A. M., Brockmann, M. M., Giusti, S. A., Maccarrone, G., Vercelli, C. A., Bauder, C. A., Richter, J. S., Roselli, F., Hafner, A.-S., Dedic, N., Wotjak, C. T., Vogt-Weisenhorn, D. M., Choquet, D., Turck, C. W., Stein, V., Deussing, J. M., & Refojo, D. (2015). Neddylation inhibition impairs spine development, destabilizes synapses and deteriorates cognition. Nature Neuroscience, 18(2), 239–251. https://doi.org/10.1038/nn.3912
- Tian, Z., Li, J., Ma, R., Li, T., Sun, Z., & Huang, S. (2023). Targeting neddylation as a novel approach to lung cancer treatment (Review). International Journal of Oncology, 62(5), 65. https://doi.org/10.3892/ijo.2023.5513
- Ying, J., Zhang, M., Qiu, X., & Lu, Y. (2018). Targeting the neddylation pathway in cells as a potential therapeutic approach for diseases. Cancer Chemotherapy and Pharmacology, 81(5), 797–808. https://doi.org/10.1007/s00280-018-3541-8
- Yavuz, A. S., Sözer, N. B., & Sezerman, O. U. (2015). Prediction of neddylation sites 18from protein sequences and sequence-derived properties. BMC Bioinformatics, 16(18), S9. https://doi.org/10.1186/1471-2105-16-S18-S9
- Milhollen, M. A., T, Traore, J. Adams-Duffy, M. P. Thomas, A. J. Berger, L. Dang, L. R. Dick, et al. (2010). MLN4924, a NEDD8-activating enzyme inhibitor is active in diffuse large B-cell lymphoma models: Rationale for treatment of NF-κB–dependent lymphoma.” Blood, 116(9): 1515–23. https://pubmed.ncbi.nlm.nih.gov/20525923/
- Liao, Y., Jiang, Y., He, H., Ni, H., Tu, Z., Zhang, S., Wang, B., Lou, J., Quan, S., & Wang, H. (2015). NEDD8-mediated neddylation is required for human endometrial stromal proliferation and decidualization. Human Reproduction, 30(7), 1665–1676. https://doi.org/10.1093/humrep/dev117
- Lewis, S. P., Willis, A. N., Johnson, A. E., Resau, J., & Burnatowska-Hledin, M. (2011). Mutational analysis of VACM-1/cul5 exons in cancer cell lines. Journal of Pathology, Microbiology, and Immunology. 119(7), 421-30. https://doi: 10.1111/j.1600-0463.2011.02747.x