Original Article - DOI:10.33594/000000822
Accepted 29 September 2025 - Published
online 24 October 2025
B Cells at the Crossroads of Cardiovascular and Hematologic Disease: Paving the Way for Novel Immunomodulatory Therapies
bDepartment of Clinical Therapeutics, Department of Medicine, School of Health Sciences, National and Kapodistrian University of Athens, 11528 Athens, Greece,
cNational Health and Lung Institute, Imperial College London, London, UK,
dDepartment of Pathophysiology, National and Kapodistrian University of Athens, School of Medicine, 11527 Athens, Greece,
eSection of Animal and Human Physiology, Department of Biology, National and Kapodistrian University of Athens, 15784, Athens, Greece
Keywords
Abstract
The interaction between the immune and cardiovascular systems is a growing field of investigation with bidirectional aspects. B cells are modulators of the adaptive and the innate immunity and they orchestrate bone marrow and spleen immune responses beyond infectious diseases. B cell regulation contributes to the pathophysiology of myocardial damage in several conditions including myocardial infarction, heart failure and atherosclerosis. In parallel, B cell-derived hematological disorders are interlinked to cardiovascular complications, including thrombosis and immunoglobulin-related cardiotoxicity. The scope of this review is to summarize the function and role of B cells as important players in myocardial and vascular adaptations to injury and as mediators of cardiovascular adverse events in hematological disorders. The primary focus is to highlight the clinical and preclinical findings regarding B cell-targeted therapies and their positive or negative impact on the cardiovascular system. A deeper understanding of B cell subpopulations, functions, and secretome could lead to targeted therapeutic interventions for cardiovascular and hematologic diseases.
1. Introduction
The adult human heart consists of a diverse population of roughly 5 billion cells which operate together within specialized tissue microenvironments (niches) to regulate each heartbeat [1, 2]. This intricate coordination is disrupted by conditions, including cardiovascular (CV) comorbidities, such as ischemic heart disease. Advances in single-cell technologies have uncovered the heterogeneity of lymphoid cell populations in both mice and humans, identifying distinct subsets with specialized functions. Previously underappreciated populations, including cardiac-resident or bone-derived hematopoietic stem cells (HSCs), mature B cells and plasma cells, can contribute to the development of heart diseases. Although, the role and function of each subset of the B lymphoid lineage remains to be fully elucidated, several data support their involvement in immune responses to myocardial injury and the increased risk of CV events observed in hematologic malignancies [3, 4].
This review focuses on the B and plasma cells bidirectional relationship with the CV system and is structured into four main sections. First, we provide an overview of the role and function of this lymphoid lineage from an immunological perspective. Next, we present a comprehensive summary of current findings regarding the involvement of B cells in the healthy and diseased myocardium. In the third section, we explore cardiovascular adverse events associated with hematologic malignancies. Finally, we examine existing pharmacological interventions targeting B cells and their effects on the CV system, aiming to suggest future directions for therapeutic strategies in both CV and hematologic diseases.
2. Maturation and functions of B cells
HSCs reside within the bone marrow and possess the capacity for self-renewal and multilineage differentiation, resulting in an approximate release of one trillion cells daily [5]. During lymphopoiesis, HSCs differentiate into common lymphoid progenitors (CLP), and lead to the origination of T and B cell lineages and natural killer cells [6]. HSCs mobilization is governed by interactions with stromal cells of the bone marrow niche, and signals such as macrophage colony-stimulating factor (CSF1), granulocyte–macrophage colony-stimulating factor (GM-CSF) and cytokines [7].
B cell development occurs along with immunoglobulin (Ig) gene rearrangements. Progenitor B cells initiate
the
combinatorial rearrangement of the heavy chain (HC)
(V, D, and J gene segments in the H chain locus)
to
differentiate into precursor B cells that express immature B cell receptors (BCR) with a surrogate light
chain
(LC)[8]. Rearrangement of the LC genes (V and J gene segments in the L chain loci) leads to their
differentiation to IgM-expressing immature B cells and to IgM+IgD+ mature resting B
cells.
Ig gene rearrangement is prone to defects resulting in the development of B cell malignancies,
immunodeficiencies or autoimmunity. Immature B cells exit the bone marrow and undergo their final
stages
of development in the spleen to form mature B cells [9] which comprise of distinct cell subsets:
follicular B
cells, marginal zone (MZ) B cells and B1 cells [10].
Follicular B cells are the dominant subset and reside within lymphoid follicles of the spleen and lymph nodes. Due to their location in follicles adjacent to T cell zones, they mount T helper-dependent responses to antigens presented by follicular dendritic cells (DCs) [11]. Follicular B cells proliferate and differentiate into antibody producing plasma cells or enter germinal centers, where affinity maturation of the BCR occurs [12].
MZ B cells, by contrast, are strategically placed in the interface between the red and white pulp in the spleen to respond to blood-borne pathogens and antigens. Their activation threshold is lower than follicular B cells, allowing IgM production in the absence of cluster of differentiation 40 (CD40)-dependent signals from follicular T helper cells. MZ B cells express polyreactive BCRs -which resemble the broad recognition of molecular patterns of toll-like receptors (TLRs)- and high levels of TLRs (like DCs, macrophages and granulocytes), allowing them to initiate low-affinity antibody responses prior to the induction of high-affinity antibody production by conventional follicular B cells [10, 13]. In mice, expression of CD1d and CD21 on MZ B cells facilitates the recognition of lipid antigens [14, 15], leading to the production of lipid-specific antibodies.
B1 cells are located in coelomic cavities such as the peritoneum and the lungs. They are spontaneously producing natural IgM antibodies without depending on thymus activation [16]. IgM is the earliest type of membrane-associated Ig expressed during B-cell ontogeny, it circulates as pentamer and is 1–2 mg/ml in the human blood. Besides host defense, IgM removes apoptotic cells and oxidized lipids from the circulation [17].
Mature B cells upon BCR activation give rise to plasmablasts —transient, proliferating cells that secrete antibodies [12]-, while plasma cells represent the terminal differentiation state of B cells and are specialized in high-rate antibody secretion. They are well distinguished from other populations due to the membrane expression of Syndecan-1 (CD138) which can also bind to fibronectin, collagen and basic fibroblast growth factor [18].
While B cells are traditionally recognized as antibody-producing cells, it is now well established that they contribute to immune regulation through a variety of mechanisms. They secrete a wide range of cytokines affecting other immune cells [19]. B cells express high levels of major histocompatibility complex (MHC) class II molecules, acting as antigen-presenting cells [20]. They recognize antigens through the BCR, internalize and process them, and subsequently present derived peptides to CD4⁺ T cells, particularly within germinal centers [20, 21], a function that has also been recognized for MZ B cells in mice [11].
B cells shape immune responses through cytokine secretion, triggered by stimuli such as TLR activation and BCR engagement. These soluble mediators exert diverse effects: B cells enhance innate immunity through the production of interferon gamma (IFN-γ), interleukin (IL)-6, and IL-17[19]; promote CD4⁺ T-cell polarization; contribute to the development of lymphoid tissues through LTα1β2[19]; mediate monocyte recruitment via C-C chemokine ligand 7 (CCL7)[22]; facilitate T-cell trafficking to inflamed tissues by secreting PEPITEM[23] and directly modulate T-cell function [24].
Conversely, certain B-cell subsets, referred to as regulatory B cells (Bregs), exert immunosuppressive functions. These cells secrete anti-inflammatory cytokines such as IL-10 contributing to immune tolerance and resolution of inflammation [19]. Bregs also promote regulatory T-cell (Treg) activity, preserve natural killer cell homeostasis, and suppress the pro-inflammatory activity of monocytes, DCs, and CD8⁺ T cells [25]. In mice, IL-10-producing Breg subsets include CD5+CD1dhi B (B10) cells, MZ B cells and their precursors, as well as plasmablasts and plasma cells [26]. In humans, comparatively fewer Breg subsets have been characterized among peripheral blood mononuclear cells (PBMCs). The immature CD19⁺CD24hiCD38hi B cell subset produces the highest levels of IL-10 along with plasmablasts, and plasma cells as in mice [26]. Their ontogeny is yet to be explored, but the balance of pro- and anti-inflammatory environments can account for the differentiation and expansion to regulatory rather than effector B cells.
Taken together, B cells originate from HSCs and undergo a stepwise developmental process initially in bone marrow and afterwards in secondary lymphoid organs, that ensures the generation of a diverse and functional BCR repertoire. Their maturation yields distinct subsets—follicular B cells, MZ B cells, and B1 cells—each occupying specialized anatomical niches and serving complementary roles in immune surveillance. Collectively, B cells are not only key producers of antibodies but also active participants in immune regulation through antigen presentation and cytokine secretion or by limiting excessive immune activation and promoting tolerance.
3. B cells in normal cardiovascular function and in disease
3.1 The presence and role of B cells in the healthy myocardium
Both B and T cells are part of the physiological immune cell landscape of the heart, even in the absence
of
disease in both humans and rodents [27]. By using single-cell and single nucleus technologies, Litviňuková
et
al. detected myeloid and lymphoid cells in humans consisting of approximately 10% and 5% of cardiac cells
in the
atrial and the ventricular regions, respectively [28]. In the same study, 8 subpopulations of lymphoid
cells
were identified including B cells and plasma cells which were shown to interact with cardiomyocytes and
fibroblasts, deducing paracrine circuits important for cardiac homeostasis [28]. Immunohistochemistry
using
antibodies against the markers CD3, CD20, and CD31 on post-mortem cardiac biopsies from individuals
without
underlying cardiac pathology, indicated that CD20⁺ B cells were present both in the interstitial
(extravascular)
and intravascular compartments, as well as across the myocardium and epicardium. The functional role of B
cells
in human heart is not fully understood, while gene expression analysis revealed that pathways related to
“B-cell
receptor signaling pathway,” “Antigen processing and presentation,” and “Cytokine-cytokine receptor
interaction”
were upregulated in cardiac B cells in comparison with peripheral B cells. Based on these, they
hypothesized
that B cells extravasate, degrade the extracellular matrix and crawl through the interstitial space where
they
communicate with other cells such as fibroblasts [29]..
In a multi-omics approach, Kanemary et al [30].
identified cardiac plasma cells in the human epicardium in two niches; one enriched in lymphatic
endothelial and
immune cells, and in a fibroblast-rich niche. Gene expression analysis revealed that IgA and IgG
expressing
cells were distinctly localized, with IgA presence being pronounced in the subepicardial region [30].
Cardiac
plasma cells expressed C-C chemokine receptor 2 (CCR2) and CXC chemokine receptor 4 (CXCR4) via which they
can
interact with endothelial cells, fibroblasts, and resident macrophages. Based on their transcriptome,
plasma
cells were predicted to interact with macrophages via B-cell activating factor (BAFF) receptors (BAFF-R).
In the
same study, endothelial, fibroblast and plasma cell interactions were mediated by transforming growth
factor
(TGF) signaling. These findings highlighted the important role of plasma cells in cardiac homeostasis,
fibrosis
and immune defense [30]. In parallel, Bermea et al. provided a functional explanation for the presence of
B
cells in the human heart [31]. They identified naïve B cells (i.e., B cells that have not been activated
or
encountered antigens) and plasma cells in the healthy heart which interacted with endothelial cells,
fibroblasts, pericytes, and smooth muscle cells via non-canonical Wnt signaling and with other B cells,
endothelial cells, and myeloid cells through PECAM1 homophilic interaction, probably participating in
transmigration or survival [31]. In an integrated analysis of existing single-cell data, de Winter et al.
identified the B cells subsets in the human heart, comprising B1 cells, naive B cells, plasma cells,
memory B
cells and Bregs, supporting the important role of B cells in cardiac homeostasis and immune tolerance
[32].
Novel omics-derived data from humans corroborate previous observations in murine hearts, showing that B
cell
subsets exist in the heart. Kalikourdis group identified B cells in the healthy murine heart via single
cell
technologies and the expression of the B220 marker [27, 29], supporting the conservation of B cell subsets
and
function in the heart between humans and mice.
Interestingly, the neonatal heart also has B cell subsets to promote cardiomyocyte proliferation [33].
During
transition to adulthood, the markets of B cells are altered and the naïve B cells have been shown to
circulate
between the heart and spleen throughout life but the functional significance of this equilibrium has not
been
identified [34].
3.2 The role of B cells in cardiovascular disease
Upon the seminal work of Adamo et al [35]. dated in 2020, we summarize the novel findings on the
contribution of
B cell subsets and Igs related to atherosclerosis, ischemic heart disease and heart failure (HF) (Fig.
1A-C).
3.2.1 Atherosclerosis
B cells have emerged as significant contributors to vascular inflammation, atherosclerosis, and
thrombosis. They
have been identified in human carotid atherosclerotic plaques [36] and their interactions with endothelial
cells, platelets, and other immune components underscore their multifaceted involvement in vascular
pathology.
Activated B cells secrete pro-inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), IL-6,
and
IL-1β, which can induce endothelial cell-activation and dysfunction and in turn, increase monocyte and
macrophage infiltration [37, 38]. This process is characterized by increased expression of adhesion
molecules,
enhanced vascular permeability, and a pro-thrombotic state, all of which are critical early events in
atherogenesis and thrombosis. Furthermore, B cell-derived cytokines can promote the recruitment and
activation
of other immune cells, amplifying vascular inflammation. B cells can influence platelet function through
the
formation of immune complexes that engage Fc gamma receptors (FcγRs) on platelets. This interaction can
lead to
platelet activation, aggregation, and the release of pro-thrombotic mediators, thereby contributing to
thrombus
formation [37]. Additionally, B cell-derived antibodies can directly bind to platelet antigens, further
modulating platelet activity and promoting a pro-thrombotic environment [39].
B cells also exhibit both protective and pathogenic roles in atherosclerosis, largely dependent on their
subset
classification. B1 cells produce natural IgM antibodies that recognize oxidized low-density lipoprotein
(oxLDL)
and apoptotic cells, facilitating their clearance, thereby exerting atheroprotective effects [40]. In
contrast,
B2 cells, comprising follicular and MZ B cells, promote atherosclerosis by producing pro-inflammatory
cytokines
and pathogenic IgG antibodies in Apoe -/- mice [41]. Bregs suppress immune responses through
the
production of IL-10 and have been implicated in the attenuation of atherosclerosis by modulating T cell
responses and reducing vascular inflammation in a mouse model of systemic lupus erythematosus (SLE) and
atherosclerosis [42].
Τhe unique B-cell subsets and their role in atherosclerosis is still under investigation. For
instance,
depletion of B2 cells using anti-CD20 antibodies or BAFF receptor antagonists has been shown to reduce
atherosclerotic lesion development in murine models [43]. A distinct subset of B1 cells—termed innate
response
activator (IRA) B cells—has been identified to respond to lipopolysaccharide (LPS) stimulation by
secreting
GM-CSF. IRA B cells are distinguished from conventional B1 cells as they express the immature B-cell
marker
CD93. IRA B cell deficiency in cholesterol-fed Ldlr⁻/⁻ mice resulted in reduced atherosclerosis
[44]. IRA
B cells can promote mature DC expansion, leading to increased IFN-γ–producing T- helper cells and a class
switch
from IgG1 to IgG2c antibodies directed against oxLDL which enhances pro-atherogenic immune responses.
Notably,
IRA B cells were found at higher levels in the spleens of patients with symptomatic cardiovascular disease
(CVD)
compared with those with asymptomatic disease, suggesting their potential role in disease progression
[44].
3.2.2 Post-ischemic myocardial injury
Cardiomyocyte necrosis and microvascular damage release intracellular components activating neutrophils,
monocytes, and macrophages [32]. CD20+ B cells in patients and B220+IgM+
B
cells in mice are recruited early after acute myocardial infarction (AMI) in the heart, contributing to
innate
and adaptive immune response to AMI via antibody-dependent and independent mechanisms.
In patients within 6 hours of AMI, B cells account for
approximately 7% of PBMCs [45]. Single-cell RNA sequencing highlighted that in both immature and mature B
cells,
chemokine signaling and immunometabolism were affected. Notably, mature B cells in patients with fibrous
plaque
rupture had markedly elevated expression of the IL-4 receptor and levels of IL-7, suggesting their
activation
and cytokine production [45]. In another study in patients, upon thrombolytic therapy for AMI, circulating
CD11b+ B1 cells correlated to the infarcted mass and B2 cells
(CD19+CD20+CD43−) remained independent predictors of left ventricular
ejection
fraction (LVEF)[46]. In parallel in patients with AMI, a pertained increase of IgG was observed that can
trigger
antigen-specific adaptive immune responses, contributing to sustained injury but these IgG epitopes
require
further investigation [47]. Tan et al, analyzed the proteins interacting with Igs in patients with AMI and
chronic stable coronary syndrome. They revealed that the Igs’ proteome landscape included factor X, which
was
validated as target of the AMI related B cell repertoire. Moreover, immunoglobulin-associated proteome
(IgAP)
was related to the pathways of complement activation and cholesterol metabolism [48].
In mice, an increased proportion of activated (CD69+) B cells in the myocardium was detected
from the
3rd day to 1 week post-AMI, and a similar increase was observed in the spleen and the
peripheral
blood at days 3 and 5. Also, at day 5 the expression of TNF-α, IL-1β, IL-6, TGF-β1, and B cell activating
factor
(BAFF) was the highest in the myocardium, spleen and peripheral blood with the most obvious myocardial
inflammatory cell infiltration [49]. Early post-AMI in both mouse and human hearts, B cells showed
increased
expression of transcription factors related to their activation. These included nuclear factor of
activated T
cells, cytoplasmic differentiation 2 (NFATC2) and MHC class II gene expression, which were normalized at
later
stages (28 days)[32]. Long-term post-AMI, maturation and memory B cell formation genes were upregulated
(i.e.,
NF-κB1 and STAT5B) in parallel with genes relative to plasma cell maturation and antibody production [32].
This
study also reported the existence of collagen-producing B cells supporting the role of B cells in fibrosis
[32].
B cell knock-out mice demonstrated decreased fibrosis in parallel to decreased gene expression levels of
cytokines and fibrosis markers (i.e. TNF-α, IL-1β, IL-6 and TGF-β1) in the myocardial tissue and the
peripheral
blood, but the exact cell origin of these cytokines was not defined [49]. Heinrichs et al. showed that B
cell
populations increased up to the 7th day post AMI in the scar area in the murine heart [50].
Heart B
cells uniquely expressed Tgfb1 and Cxcr5 and Ccr7 receptors compared to B cells from
the
lymph nodes. Antibody-mediated neutralization of CXCL13 was performed and when Cxcr−/−
mice were employed, B cell recruitment and cardiac Tgfβ1 expression was
ameliorated [50]. Ultimately these strategies had a neutral effect on cardiac function and morphology post
AMI,
questing the role of B cells as a source of TGF-1 mediated fibrosis.
Clonal expansion of B cells in lymph nodes and natural IgM infiltration in the heart, as observed in mice,
[50]
could account for a future strategy to target B cells in AMI. Natural IgM antibodies contribute to the
myocardial infarct size. Mice bearing either a selective (Cr2−/−) or total
deficiency
(RAG-1−/−) in IgM demonstrated reduced infarct size which was comparable to wild
type
mice when IgM was injected [51]. Besides the heart, the role of splenic MZ B cells was highlighted in AMI
mice
with permanent ligation. Sun et al. demonstrated that miR21 or Hif1a-specific deletion from MZ
cells
ameliorated cardiac function [52]. They also proved that miR21/HIF-1α signaling in MZ B cells results in
TLR-dependent CCL7 expression which leads to inflammatory monocyte recruitment in the ischemic myocardium
[52].
Despite several reports of the contribution of B cells in myocardial damage, specific B cell subsets have
favorable effects on cardiac function. Intramyocardial injection of B-lymphocytes early post-infarction in
rats
resulted in preserved LVEF and amelioration of apoptosis [53]. This population was CD45RA+ and
comprised mainly immature and mature B cells [53], suggesting that non-activated immature B cells can have
a
protective role. In parallel, B cells can interact with neutrophils leading to their phagocytosis from
macrophages in vitro and in vivo, which overall confers a cardioprotective effect [54].
Bregs possess anti-inflammatory effects, although the exact mechanism mediating this effect is not
fully
elucidated [55]. In a small cohort of patients after AMI, circulating levels of
CD24hiCD38hi Bregs, but not of CD24+CD27+ B10 cells,
were
reduced compared to stable angina pectoris, suggesting the role of Bregs in inflammation upon plaque
rupture and
thrombi formation [56]. In the pericardial fat of murine hearts, IL-10 producing Bregs attenuated monocyte
recruitment and IL-10 depletion led to exacerbated myocardial injury and function [57]. A therapeutic
strategy
that was shown to increase Bregs subsets is via low doses of IL-2 in patients with type 1 diabetes or
BACH2
suppression in murine and human isolated B cells [58].
Hu et al. reported that AMI stimulates glucocorticoid release from the neuroendocrine system, inducing
sodium-hydrogen exchanger-1 (NHE1)-mediated autophagic death of bone marrow B cells, accompanied by
reduced
lineage commitment, activation, and antibody production, while splenic B cells expanded [59]. In the same
study,
the authors suggested that the sodium glucose co-transporter-2 (SGLT-2) inhibitor empagliflozin which has
cardioprotective effects [60–62], preserves the bone marrow naïve B cell populations [59]. These results
are in
line with the myocardial structural and functional improvements 72 hours post-AMI by the infusion of bone
marrow- (but not spleen or blood) derived B cells into C57BL/6J mice prior to MI induction [59].
Empagliflozin
reduced myocardial infiltration of Ly6Chi inflammatory monocytes and neutrophils in AMI mice
[60],
attenuated the percentage of myeloid cells in the spleen, and in parallel reduced cardiac GM3 gangliosides
which
induce immune cell recruitment in a TLR-dependent manner [63]. Empagliflozin also reduced circulating
white
blood cell-counts in diabetic-AMI patients [60], altogether bringing the immune-heart axis in the
forefront of
the suggested cardioprotection by SGLT-2 inhibitors.
3.2.3 Heart failure
In the failing heart of patients, microarray data reported increased myocardial infiltration of memory B
cells,
producing antibodies, cytokines and chemokines involved in the development of HF[64]. In the peripheral
blood,
increased CD19+ or TNF-producing cells have been reported for HF patients [29]. The functional
role
of cardiac B cells has been investigated in the context of dilated cardiomyopathy (DCM) and arrhythmogenic
right
ventricular cardiomyopathy (ARVC) in humans by the group of Bermea et al [31]. In ARVC, the interaction of
B
cells with other populations was reduced compared to healthy hearts. In DCM, an increased interaction with
macrophages, monocytes, cardiomyocytes, and endocardial cells was observed, possibly through the
activation of
the PI3K pathway. B cell communication with fibroblasts, epicardial fat cells, and other stromal cells was
increased via the interaction of extracellular matrix proteins such as laminin, collagen, and fibronectin
with
CD44 on B cells. In the same study, eosinophils interacted with B cells through the CD44/CD74 complex,
which
activates NF-κB signaling, suggesting that B cells play a role in eosinophil activation in DCM.
Rats with heart failure with reduced ejection fraction (HFrEF) of ischemic origin have elevated mature IgG
isotypes in their circulation targeting myosin, troponin or complement (C)3. Similarly, IgGs and C3 heart
depositions were evident in HF patients in the perivascular and interstitial space [65]. Martini et al.
suggested that cardiac B cells are activated and expanded in a murine model of pressure overload
(transverse
aortic constriction, TAC)[66]. B cells clustered together but failed to organize in follicles despite the
expression of Cxcr5. The role of auto-antibodies is highlighted by the study of Smolgovsky et al
[67]. in
which, TAC results in protein modifications with isolevuglandins (lipid peroxidation products) and in B
cell
responses with the production of anti-isolevuglanin antibodies. These effects were not replicated in heart
failure with preserved ejection fraction (HFpEF). Recently, Feng et al [68]. in a TAC mouse model
identified
that splenic IgE+ B cells are elevated due to the reduction of CD23, which negatively regulates
IgE+ production. A key mediator for this process was lactotransferrin which is released by the
heart
[68]. HF patients have also increased IgE levels.
The role of inflammation in HFpEF patients is appreciated based on conventional markers (i.e., hs-CRP,
IL-6)
that predict morbidity and mortality, but whether this immune activation is directly related to B cell
functions
is ill-defined.
The main driver of HFpEF is hypertension. Guzik at al [69]. reviewed the immunological aspects observed in
hypertension. Angiotensin II infusion leads to increased splenic B cells and plasma cells in mice, in
parallel
with augmented circulating and aortic IgG. B cell depletion or BAFF receptor deficiency attenuated
hypertension,
aortic accumulation of macrophages and stiffening [70]. However, the role of Ig production has been
debated
since global B cell or IgM deficiency did not result in the attenuation of hypertension [71]. Yet in an
angiotensin‐II‐induced HF mouse model, B‐cell absence resulted in preservation of the cardiac function,
attenuation of hypertrophy and collagen deposition in parallel with reduced IgG deposition [72].
In patients with diastolic dysfunction, elevated circulating levels of IgG1 and IgG3 were detected which
corroborated with cardiac remodeling [73]. HSC proliferation via metabolic regulation of neighboring
myeloid
cells has been identified as a driver of HFpEF, leading to the modulation of macrophage adhesion molecule
Vcam1 in a murine 2-hit model of HFpEF that incorporates obesity and hypertension [74].. Of
note,
β2-microglobulin, a component of MHC class I which is elevated in patients with B cell malignancies, has
been
found elevated also in patients with pulmonary hypertension and HFpEF predicting disease severity [75].
HFpEF
was also associated with decreased lympagiogenesis and lymphatic endothelial cells in mice and patients
[76].
The possible relation of these findings to B cell regulation remains to be elucidated.
In summary, in the healthy myocardium, B cells, including naïve, memory, B1, plasma cells, and Bregs, reside in interstitial, intravascular, myocardial and epicardial compartments, interacting with endothelial, stromal cells and macrophages that support tissue surveillance, yet more roles are soon to be clarified. In cardiovascular disease, B cells exert subset- and condition- specific effects. In atherosclerosis, B1 cells and Bregs provide atheroprotection, while B2 and IRA B cells promote inflammation, pathogenic IgG responses and a pro-thrombotic environment. (Fig. 1A) In post-ischemic myocardial injury, B cells are early recruited and activated, contributing to cytokine production, fibrosis and antibody-mediated tissue damage, while cardioprotection via Bregs and immature B cells has been reported (Fig. 1B). In heart failure, B cells participate in maladaptive remodeling through cytokine production, autoantibody generation, and interactions with cardiomyocytes, eosinophils, macrophages, and stromal cells (Fig. 1C). Their exact role in HFpEF remains less clearly defined. Overall, B cells serve as a promising target for therapeutic modulation in cardiovascular disease since B cell depletion or modulation could mitigate inflammation, hypertrophy, and functional decline.
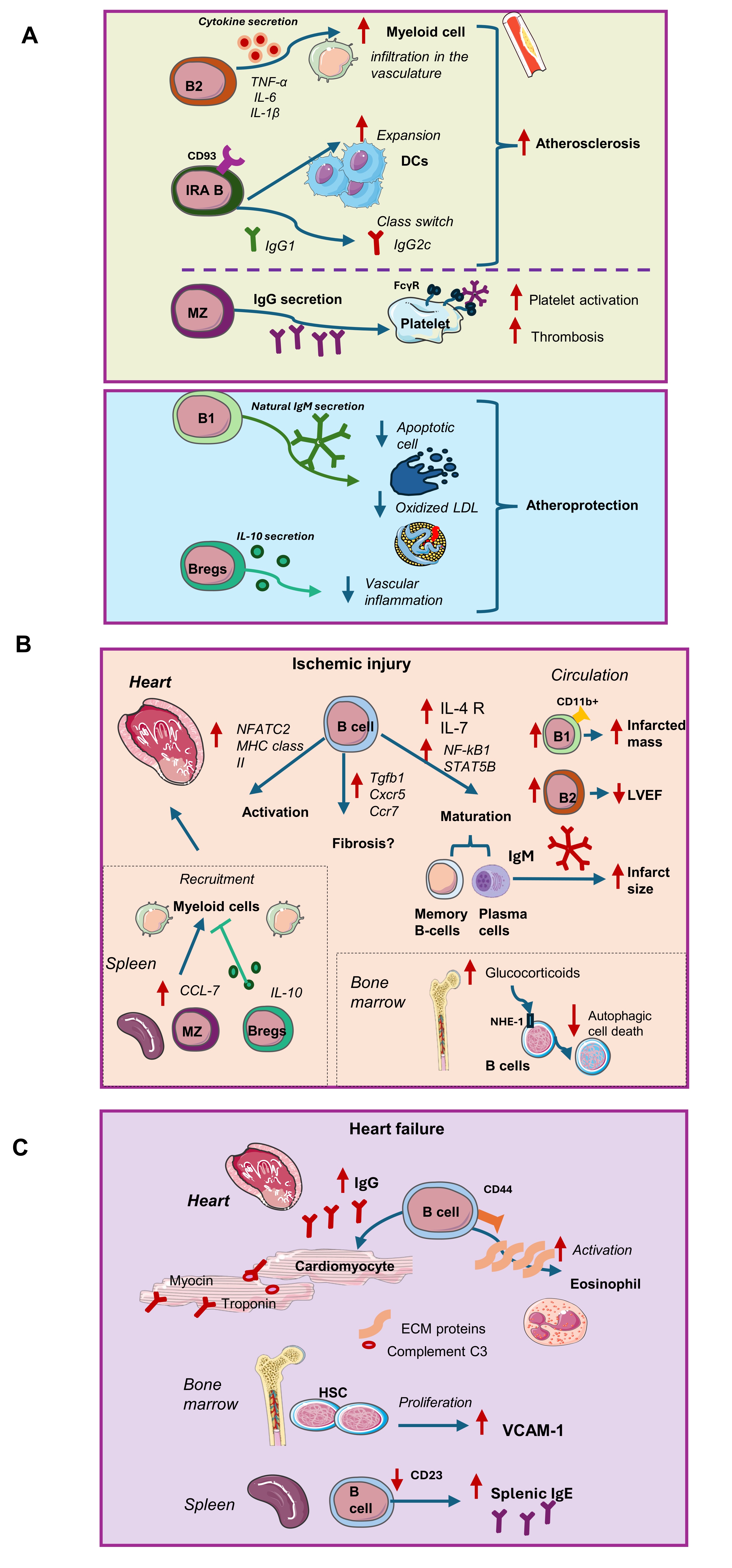
Fig. 1: Molecular mechanisms suggested for the interplay of B cells in cardiovascular disease. This Fig. summarizes the proposed mechanisms by which B-cell subsets contribute to cardiovascular pathology. A) In atherosclerosis, splenic B2 cells release proinflammatory cytokines that promote myeloid cell infiltration, increase vascular permeability, and amplify inflammation. IRA B cells expand DCs and enhance IgG class switching, whereas MZ B cells promote thrombosis through IgG-mediated platelet activation. Τhese subsets may promote atherosclerosis. Conversely, B1 cells exert atheroprotective effects by producing natural IgM that recognizes oxLDL and apoptotic cells, while Bregs secrete IL-10, attenuating vascular inflammation. B) In ischemic injury, CD20⁺ B cells are rapidly recruited after AMI, expressing genes linked to activation, fibrosis and differentiation into plasma and memory B cells. Antibody production (IgM, IgG) contributes to infarct size, partly through complement activation. Circulating CD11b⁺ B1 cells correlate with larger infarcts, whereas B2 cells are associated with LVEF. splenic MZ B cells secrete CCL7, driving myeloid cell recruitment to the infarcted heart, an effect counteracted by IL-10–producing Bregs. Bone marrow B cells undergo glucocorticoid-driven autophagic death mediated by NHE1. C) In heart failure, HSC proliferation results in macrophage VCAM-1 upregulation. B cells interact with eosinophils via CD44, and IgG antibodies against myosin, troponin, or complement C3 are detected in the heart. Reduced CD23 expression increases splenic IgE levels, further amplifying inflammation. B regs: B regulatory cells; CCL/CCR: C-C chemokine ligand/receptor; CXCR:C-X- chemokine receptor, DCs: dendritic cells; ECM: extracellular matrix; FcγRs: Fc gamma receptors; HSC: hematopoietic stem cells; Ig: immunoglobulin; IL: interleukin; IRA: Innate response activator; LVEF: Left ventricular ejection fraction; MZ: marginal zone B cells; NHE1: sodium-hydrogen exchanger-1; NFATC-2: nuclear factor of activated T cells, cytoplasmic differentiation 2; oxLDL: oxidized LDL, TNF-a: tumor necrosis factor a, VCAM-1:vascular adhesion molecule -1.
4. Cardiovascular disease in B cell-related hematologic malignances
Most B-cell hematologic malignancies are linked with increased CV risk due to a complex interplay of disease, treatment and patient-related factors.
Multiple myeloma (MM), a plasma cell malignancy with monoclonal protein secretion, predominantly affects
the
elderly—who are already at higher baseline CV risk. Up to 7.5% of MM patients experience CV events such as
HF,
arrhythmias, or ischemia [77, 78]. MM is frequently associated with CV comorbidities,
influenced by both disease-related and treatment-related factors. For instance, anemia and renal
dysfunction are
independent predictors of CVD, and hypercalcemia can predispose to arrhythmias.
Venous thromboembolism (VTE) is a common and serious complication associated with MM or its treatment. VTE rates range from 4% - 16%[79] and MM patients have a 9-fold increased risk of VTE compared to the general population. In patients with MM, monoclonal protein has been linked to hyperviscosity, hyperfibrinogenemia, decreased protein C or S activity, impaired fibrinolysis and interferes with coagulation. In parallel, elevated IL-6 and TNF-α, as well as plasma cell-derived extracellular vesicles cause endothelial cell and platelet activation [80]. Therapy-related factors contribute significantly to the risk observed, like corticosteroids, proteasome inhibitors (PIs)[81, 82], high-dose melphalan, immunomodulatory drugs (IMiDs) [83, 84] and novel agents such as cellular therapies [85, 86],. IMiDs in particular, increase significantly the risk of thrombosis [87, 88].
Waldenström’s macroglobulinemia (WM), defined as a lymphoplasmacytic lymphoma with IgM paraproteinemia, is associated with a 2–4 fold elevated VTE risk [89] but not with arterial thrombosis risk. IgM-related hyperviscosity impairs the microcirculation and promotes thrombosis through red cell aggregation and clotting factor interference [90, 91]. Inflammatory cytokines and elevated FVIII levels exacerbate the prothrombotic environment [92, 93].
Monoclonal gammopathy (MG) is characterized by the presence of a monoclonal protein produced by low-grade plasma or B-lymphocytic clone. Monoclonal gammopathy of undetermined significance (MGUS) is present in 3.2% of Caucasians aged 50 years [94, 95], an incidence that increases with age [96]. Many studies have demonstrated an association between MGUS, CV risk [97, 98] and VTE risk [97], but the link is mostly epidemiological. The recent iSTOPMM study evaluated patients with “screened”, rather than “clinical MGUS” and confirmed a 1.3-fold increased VTE but not arterial thrombotic risk [99]. Studies on biomarkers suggested possible shared biologic features between MM and MGUS like platelet activation and hypercoagulability [100, 101]. The term Monoclonal Gammopathy of Thrombotic Significance (MGTS)[102] has been introduced to describe patients presenting with clinically significant thrombotic complications linked to the M-protein with uncommon thrombogenic properties, such as anti-platelet factor 4 activity but otherwise no overt malignancy [103, 104].
Excessive production of LCs by plasma cells and their deposition as amyloid fibrils in various organs comprises the clinical entity of light chain amyloidosis (AL), which is a separate but related condition that results in cardiac involvement. In AL amyloidosis, a low-grade clonal plasma or B cell population produces amyloidogenic free LCs that aggregate to form insoluble fibrils, which deposit in tissues causing organ dysfunction [105]. Over 75% of patients have cardiac involvement at diagnosis [106]. Even in the prefibrillar state, the amyloidogenic LCs (λ subtype in 80%) induce cardiotoxicity and increase cellular oxidative stress in human heart cells [107]. Amyloid infiltration results in ventricular wall thickening, diastolic dysfunction (initially with preserved LVEF), and poor atrial contractility increasing the risk of thromboembolic complications, and atrial or ventricular arrhythmias. LCs cause diastolic dysfunction in isolated murine hearts [108], and result in direct impairment of cardiomyocyte contractility and calcium handling in adult rat ventricular cardiomyocytes [109]. Cellular abnormalities involved in LCs’ cardiotoxicity include impairment of lysosomal activity and autophagy in isolated rat cardiomyocytes and in zebrafish [110, 111]. In vitro, the activation of a non-canonical p38α MAPK pathway has also been related to LC cardiotoxicity [112] along with mitochondria dysfunction and integrity by interacting with mitochondrial targets, such as OPA-1[113]. The role of inflammation in LC-mediated toxicity and amyloidosis has been also suggested [114], while novel mechanisms of cardiotoxicity are currently under investigation [115].
Non-Hodgkin lymphoma (NHL) encompasses a heterogeneous group of hematological B-cell neoplasms with varying clinical and pathological features [116]. Diffuse large B cell lymphoma (DLBCL), follicular lymphoma (FL) and chronic lymphocytic leukemia (CLL) are the most common subtypes. Other B cell malignancies include Hodgkin lymphoma (HL), with germinal center B lymphocytes that underwent transformation during maturation, and B cell acute lymphoblastic leukemia (ALL), an aggressive malignancy of B lymphoblasts. VTE is prevalent among B-cell hematologic malignancies [117]. The risk depends on patient-related comorbidities, immobility, previous VTE and treatment-related factors. There is an association between high-tumor burden and aggressive cancer histology [92, 118]. Stark et al [119]. demonstrated in mice and humans that reduced blood flow velocity allows IgM to bind to the endothelium, resulting in surface exposure of P-selectin and vWF, initiating platelet recruitment to the veins. This process is followed by platelet activation, promoting the deposition of IgG depending on fibrin and chondroitin sulfate-A but irrespectively of the antigen specificities of the IgG. This vicious circle of platelet activation leads to leukocyte recruitment and clot formation [119]. We need more data to understand the underlying pathogenetic mechanisms which contribute to an inherently prothrombotic state in clonal B-cell malignancies.
Treatment advances over the last decades have significantly improved the survival of these patients, making the impact of toxicities and comorbidities increasingly evident. The risk and prevalence of CV comorbidities and associated mortality is significantly higher in patients with NHL and HL compared to the general population [120, 121]. NHL patients have up to a 14-fold higher risk of CV mortality [122]. The risk is higher for more aggressive NHLs. Multiagent chemotherapy HSC transplantation and mediastinal radiation [123] also induce significant cardiotoxicity.
Clonal haematopoiesis, defined as somatic mutations and mosaic chromosomal alterations in blood cells including lymphocytes, is currently investigated as a primer of CVD (reviewed in [124–126]).
Overall, B-cell hematologic malignancies are closely intertwined with cardiovascular pathology, not only through treatment-related toxicities but also via intrinsic disease mechanisms such as hyperviscosity, hypercoagulability, inflammation, and amyloid LC–mediated cardiotoxicity.
5. Immunomodulatory therapies targeting B cells and their cardiovascular effects
5.1 Cluster of Differentiation (CDs) - Cell Surface Antigens
Multiple B-cell–depleting therapies, such as monoclonal antibodies (mAb) and bispecific T-cell engagers
(BiTEs),
are approved for the treatment of hematologic malignancies and autoimmune diseases. These agents can
influence
the CV system in both beneficial and adverse ways (Fig. 2).
Rituximab (anti-CD20) is a chimeric mAb targeting the CD20 antigen on mature B lymphocytes [127], but not
on
stem cells or plasma cells, offering a targeted approach to modulate B-cell-driven pathology. Rituximab
induces
the selective temporary depletion of CD20+ B cells, via antibody-dependent cell-mediated
cytotoxicity
(ADCC) and complement-dependent cytotoxicity [128]. It has been approved for the treatment of lymphoma,
rheumatoid arthritis (RA), and ANCA-associated vasculitis, showing favorable effects during off-label use
for
several immune-mediated diseases such as SLE[128]. Within 48 h of rituximab infusion, adverse cardiac
events
such as acute HF or AMI emerge, due to cytokine release; nevertheless, long term use of the antibody is
beneficial for patients with chronic myocarditis or a heart transplant [129].
In both preclinical and clinical investigations, the administration of CD20-targeted agents has shown
positive
impact on cardiac remodeling and HF. In specific, in a TAC myocardial hypertrophy model, Ma et al. showed
that
rituximab significantly enhanced heart function, reduced myocyte hypertrophy, fibrosis, and oxidative
stress
[130]. Similarly, in murine models of AMI, anti-CD20 antibody treatment, targeting murine CD20, improved
cardiac
function and decreased adverse ventricular remodeling [22]. B cell depletion can be beneficial for
specific
types of myocardial injury such as myocarditis. Tschope et al. observed that six patients with DCM who did
not
respond to initial immunosuppressive treatment, had CD20+ B cell infiltration in endomyocardial
biopsies and responded to rituximab infusions, with improved cardiac function [131].
Preclinical data and case reports encouraged further clinical investigations. The RITA-MI (Rituximab in
Patients
With ST-Elevation Myocardial Infarction) study was the first translational phase I/IIa trial to
hypothesize the
cardioprotective effects of anti-CD20-targeted therapies in humans. Rituximab resulted in significant B
cell
depletion—more than 95% within 30 minutes of infusion—with sustained and dose-dependent effects on B-cell
repopulation, lasting up to six months, while it was well tolerated, with no serious side effects. The
200-mg
dose demonstrated the increase of transitional B cells at 6 months, which are known to have a regulatory
phenotype, while with the 1000-mg dose, the B-cell compartment was significantly depleted. Ig levels
remained
unaffected throughout the follow-up period [132]. Currently, the CV effects of rituximab are investigated
via
the RITA-MI-2 study (RITA-MI-2; https://www.clinicaltrials.gov; unique identifier: NCT05211401) aiming to
determine if post–AMI B cell depletion yields measurable clinical benefits in humans [133]. In support of
the
above, patients with pemphigus treated with rituximab had improved long-term CV and metabolic outcomes
when
compared to those receiving azathioprine or mycophenolate mofetil (MMF). Rituximab treatment significantly
reduced the risk for AMI, stroke, peripheral vascular disease, hypertension, hyperlipidemia, type 2
diabetes,
obesity, and osteoporosis, with no increase in all-cause mortality, providing evidence for the superior CV
and
metabolic safety of rituximanb over conventional immunosuppressants, particularly in patients with
underlying
risk factors [134]. Regarding the effect of CD20-targeting in atherosclerosis progression, the hypothesis
remains to be established via RITA-MI 2.
Beyond CD20, CD38 is another B-cell differentiation antigen, which serves as both a multifunctional
receptor and
an ectoenzyme (extracellular enzyme). It is highly expressed on the surface of B cells, including
plasmablasts
and plasma cells, as well as in non-hematopoietic tissues, such as neurons, endothelial cells and
cardiomyocytes
[135].
In MM and AL amyloidosis, mAbs targeting CD38 exploit its overexpression on malignant plasma cells to
trigger
cytotoxic effects [136, 137]. CD38 has emerged as a potential therapeutic target for CVDs, due to its
involvement in the pathogenesis of AMI, atherosclerosis, cardiac arrhythmias, myocardial hypertrophy and
pulmonary hypertension [138]. As the primary NAD⁺/NADP⁺-degrading ectoenzyme [139], CD38 plays a crucial
role in
the regulation of NAD⁺ homeostasis, modulating energy metabolism and calcium signaling through the
CD38/cyclic
adenosine diphosphate ribose/Ca2+ signaling pathway [135, 140]. This multifaceted role
underscores
potential therapeutic implications in ageing, metabolic disorders, and CVD. Supporting this, experimental
studies have shown that CD38 inhibition, via miR-499a-5p-mediated downregulation in an vitro model of
hypoxia/reoxygenation or pharmacological agents such as thiazoloquin(az)olin(on)e 78c in an ex vivo model
of
myocardial ischemia-reperfusion (IR) injury, can reduce myocardial injury and endothelial damage following
ischemia [141, 142]. Additionally, CD38 deficiency has been associated with reduced oxidative stress in
the
cardiac tissue from CD38-/- mice fed with high fat diet [143].
In clinical practice, daratumumab, a human anti-CD38 antibody, is approved as monotherapy or in
combination with
standard-of-care regimens for newly diagnosed or relapsed/refractory (R/R) MM and AL amyloidosis patients
[144].
Multiple clinical studies have reported Daratumumab’s potential to exert a cardioprotective effect.
In
the phase III CANDOR trial, in patients receiving daratumumab on top of carfilzomib and dexamethasone, the
incidence of grade ≥ 3 cardiac failure was lower [136, 145]. In the phase III ANDROMEDA trial, in patients
with
newly diagnosed AL amyloidosis the addition of daratumumab to bortezomib, cyclophosphamide, and
dexamethasone
(D-VCd) resulted in higher rates of hematologic complete response in the overall study population and
across all
cardiac stages [137]. Although, HF was one of the most common grade 3 or 4 adverse events, a post hoc
analysis
revealed, that the exposure-adjusted incidence rate for cardiac events was actually lower with D-VCd than
VCd
(median exposure 13.4 and 5.3 months, respectively)[137, 146]. The mechanism behind this speculated
cardioprotective effect is not fully understood but could be related to the inhibition of the
ectoenzymatic
activity of CD38, which restores metabolic disequilibrium and calcium homeostasis in the cardiac tissue.
Isatuximab, another anti-CD38 mAb used in the treatment of MM, was initially associated with hypertension
and
arrhythmias, including atrial fibrillation [129, 147]. However, in a phase II trial, dyspnea was reported
as the
only cardiac-related event with no significant difference between patients receiving isatuximab as
monotherapy
or in combination with dexamethasone [148]. Similarly, the phase III IKEMA trial isatuximab showed no
significant difference in the incidence of cardiac events [149].
Inotuzumab ozogamicin, an anti-CD22 mAb, is approved for the treatment of R/R CD22-positive B-cell
precursor
ALL. CD22 is an inhibitory receptor expressed on B cells, and its targeting has shown immunomodulatory
effects
beyond oncology [150]. Specifically, in a mouse model of angiotensin II-induced nonischemic
cardiomyopathy,
administration of an anti-CD22 antibody attenuated cardiac hypertrophy and myocardial fibrosis,
accompanied by
reduced expression of pro-inflammatory cytokines such as IL-1β and TNF-α, decreased myocardial IgG
deposition,
and apoptosis [151]. Despite these promising preclinical findings, there are currently no clinical data
supporting the CV benefit of inotuzumab in humans. On the contrary, veno-occlusive disease and QT interval
prolongation, has been reported in patients receiving inotuzumab treatment [129].
Finally, abatacept, a cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) Ig, is used for the
treatment
of RA. It is designed to target CD80/CD86 on antigen-presenting cells, such as B cells, blocking their
interactions with both the costimulatory receptor CD28 and the inhibitory receptor CTLA-4 on T cells,
thereby
limiting T-cell activation and immune responses [152, 153]. In B cells, this results in the promotion of
Bregs
functions by enhancing their ability to produce IL-10 and TGF-β[154, 155]. Abatacept reduced the CV risk
when
compared with a TNF inhibitor [156, 157] and decelerated atherosclerosis progression [158, 159].
Moreover,
it showed cardioprotective effects in a mouse model of myocardial IR injury [160].
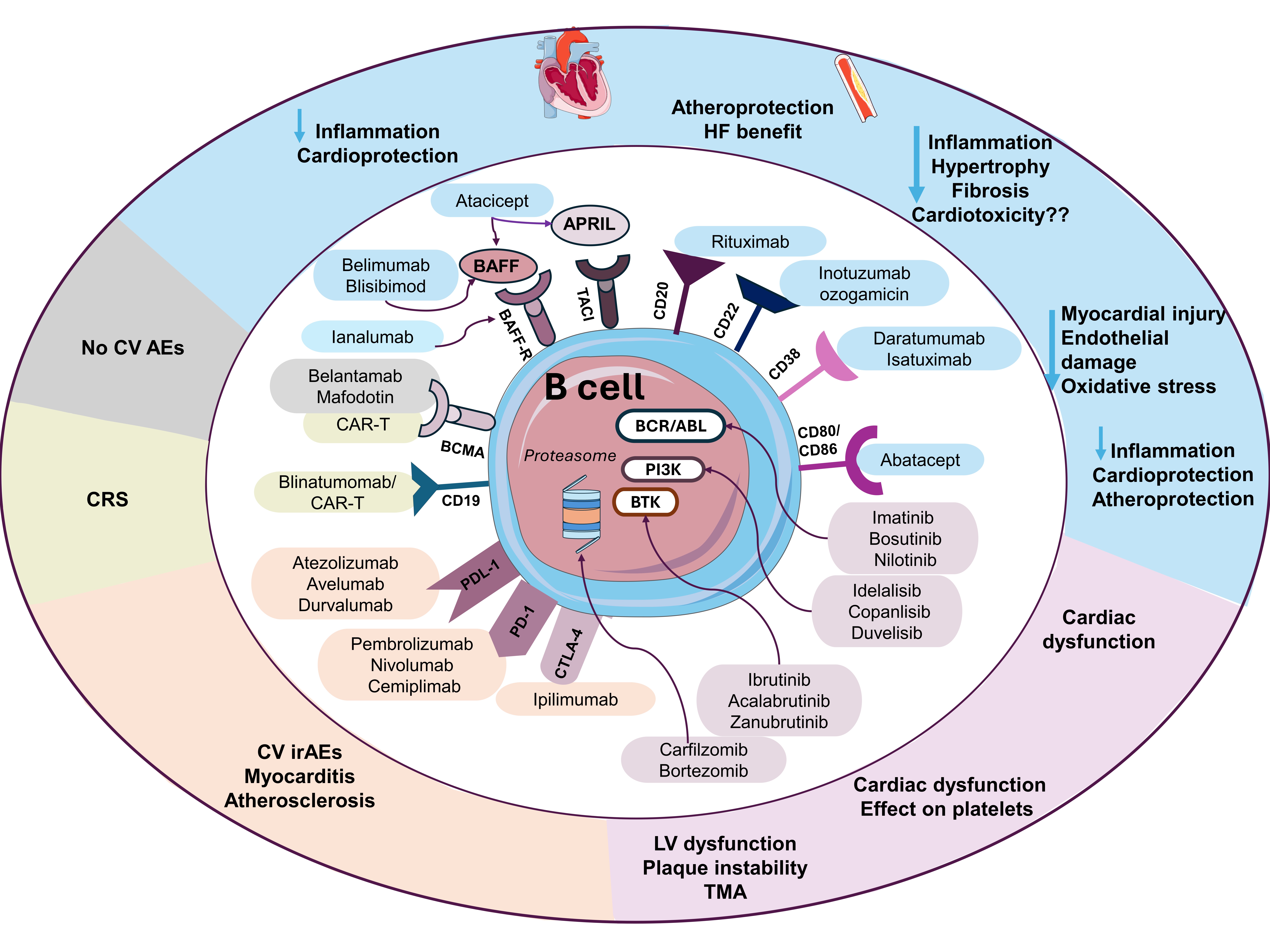
Fig. 2: B-cell–targeted therapies and their impact on the cardiovascular system. This Fig. summarizes approved B-cell–targeted therapies, their molecular targets, and their reported cardiovascular effects. These agents, developed for hematologic malignancies and autoimmune diseases, act on extracellular B-cell antigens such as CD20 (rituximab), CD22 (inotuzumab ozogamicin), CD38 (daratumumab, isatuximab), CD80/86 (abatacept), and CD19 (blinatumomab, CAR-T cells). They also target regulators of B-cell survival, including BCMA (belantamab mafodotin, CAR-T cells), BAFF (belimumab, blisibimod, atacicept), and BAFF-R (ianalumab), as well as immune checkpoints such as CTLA-4 (ipilimumab), PD-1 (pembrolizumab, nivolumab, cemiplimab), and PD-L1 (atezolizumab, avelumab, durvalumab). In addition, intracellular signaling pathways are targeted by BTK inhibitors (ibrutinib, acalabrutinib, zanubrutinib), PI3K inhibitors (idelalisib, copanlisib, duvelisib), BCR/ABL inhibitors (imatinib, bosutinib, nilotinib), and proteasome inhibitors (carfilzomib, bortezomib). By modulating B-cell function and survival, these therapies exert both beneficial and adverse cardiovascular effects, as demonstrated in preclinical and clinical studies. Beneficial effects (blue shading) include reduced inflammation, cardioprotection, and potential benefit in heart failure and atherosclerosis. In contrast, adverse effects (orange/pink shading) encompass cardiovascular irAEs, including myocarditis, atherosclerosis, cardiac dysfunction, plaque instability, TMA, and platelet dysfunction. CRS (green shading), typically observed with CAR-T therapies and BiTEs, indirectly affects the cardiovascular system, while some therapies show no clear cardiovascular adverse events, or their impact remains uncertain (grey shading). By organizing drugs according to their molecular targets and linking them to cardiovascular outcomes, the figure underscores the dual role of B-cell–targeted therapies as both potential mediators of cardioprotection and contributors to cardiotoxicity CV: Cardiovascular; CRS: Cytokine release syndrome; HF: Heart failure; AEs: Adverse events; irAEs: immune-related adverse events; LV: Left ventricular; TMA: Thrombotic microangiopathy; CD: Cluster of differentiation; BAFF(-R): B-cell activating factor (Receptor); BCMA: B-cell maturation antigen; TACI: transmembrane activator and calcium-modulator and cyclophilin ligand interactor; APRIL: proliferation-inducing ligand; PD-1: programmed cell death protein- 1; PD-L1: programmed cell death ligand- 1; CTLA-4: cytotoxic T lymphocyte-associated antigen 4; BTK: Bruton’s tyrosine kinase; PI3K: Phosphatidylinositol-3-Kinase; BCR/ABL: Breakpoint cluster region/Abelson tyrosine kinase
5.2 Receptors
B-cell maturation antigen (BCMA) is a receptor expressed on mature B cells and plasma cells, interacting
with
BAFF and proliferation-inducing ligand (APRIL)[161]. Belantamab mafodotin, a BCMA-directed drug-antibody
conjugate linked to a cytotoxic agent maleimidocaproyl monomethylauristatin F (mcMMAF), is approved for
the
treatment of R/R MM[162] and is currently under investigation in clinical trials (EMN27-NCT04617925) as a
potential therapy for R/R AL amyloidosis. This antibody functions by inducing ADCC against myeloma cells.
Both
clinical trials and real-world data indicate a good safety profile with no CV adverse events reported
[163–165].
Apart from BCMA, BAFF-R is present on the surface of mature B cells and plasma cells and has a potent
selective
affinity for BAFF, inducing downstream signaling pathways that lead to B-cell survival [166]. Preclinical
studies propose that targeting the BAFF pathway can have a potential therapeutic benefit CVDs,
particularly AMI
and atherosclerosis. In a mouse model, BAFF mAb-mediated B2 cell depletion hampered inflammation and
CCL7-mediated monocyte mobilization to the ischemic heart and was associated with decreased myocardial
injury
and improved cardiac function post-AMI[22]. Moreover, Ldlr−/− and Apoe−/− mice with
genetic or pharmacological depletion of BAFF-R exhibited a significant reduction in the B2 cell population
and
decreased lesion formation [167–169]. However, BAFF-targeted antibody in Ldlr−/− and
Apoe−/− mice aggravated the size and complexity of atherosclerotic lesions despite depletion of
B2
cells [170].
Currently, there are a few drugs in use or under investigation targeting the BAFF-BAFF-R axis, mostly for
hematological malignancies and auto-immune disorders. Among them, belimumab, a BAFF mAb, is the only one
approved for treatment-resistant SLE[133]. In clinical practice, belimumab improves the lipid profile with
an
increase in HDL[171, 172] and has been associated with the enhancement of cardiac function in a patient
with SLE
and HFpEF[173]. Other investigational agents include blisibimod, a selective antagonist of BAFF,
ianalumab,
a mAb to BAFF-R, and atacicept, a transmembrane activator and calcium-modulator and cyclophilin ligand
interactor (TACI) recombinant fusion protein that binds both BAFF and APRIL. Thus far, no significant CV
adverse
events have been reported for these drugs [133].
Recently, B cells and their role in the tumor microenvironment are gaining growing attention. In this
direction,
various tumor-associated immune checkpoints (ICs) on the surface of B cells are identified and observed to
be
upregulated in the tumor [174]. These include T cell immunoglobulin and mucin domain-containing protein
(TIM-1)[175], CTLA-4[176], tumor-infiltrating T cell immunoreceptor with immunoglobulin and ITIM domain
(TIGIT)[177], programmed cell death protein-1 (PD-1)[178] and programmed cell death-ligand 1 (PD-L1)[179].
Aberrant B-cell IC signals disrupt the function of B cells per se, promoting the release of IL-10 and
modulating the cellular functions of antigen-presentation, co-stimulation and memory. They also
affect
the tumor-killing functions of CD4+ T cells, CD8+ T cells, and Tregs, leading to
tumor
immune escape [174, 175].
Blocking ICs on B cells is advantageous for boosting anti-tumor immune response and suppressing tumor
progression [174]. To date, antibodies targeting four ICs, namely CTLA-4 (ipilimumab), PD-1
(pembrolizumab,
nivolumab, cemiplimab), PD-L1 (atezolizumab, avelumab, durvalumab), and lymphocyte activation gene 3
(LAG-3)
(relatlimab), have been approved for the treatment of several malignancies, such as melanoma, non-small
cell
lung cancer, classical HL, either as monotherapy or as adjuvant therapy [180].
Clinically, immune checkpoint inhibitors (ICIs) have been associated with immune-related adverse events
(irAEs),
which encompass rare but severe CV complications, such as myocarditis, cardiomyopathy, pericardial
disease,
arrhythmias, and atherosclerosis [181]. Underlying cardiotoxicity mechanisms have been proposed, including
increased systemic activation of T cells and release of pro-inflammatory cytokines [182] and especially
IL-17A[183], endothelial activation and microvascular coronary endothelial dysfunction [184, 185] and
cross-reactivity between tumor-infiltrating T cells and common antigens in cardiac and endothelial cells
[186].
However, only scarce data exist about the role of B cells. A few studies have described possible
mechanisms of B
cell-mediated toxicity, either through the activation and expansion of B autoreactive populations due to
activation of autoreactive clones by ICIs, recognition of neoantigens homologous to non-cancer antigens,
or
modified cytokine expression profiles [187]. Interestingly, increased activation of B cell and plasmablast
levels as well as the presence of autoreactive antibodies have been observed in ICI-treated patients at
increased risk of irAEs [187, 188].
5.3 Interaction with other cells
Chimeric Antigen Receptor T-cell (CAR-T) and BiTE therapies have transformed the treatment landscape for
hematological malignancies such as ALL, B-cell lymphomas and MM. CV toxicities have emerged as an
increasingly
recognized cause of treatment-related morbidity and mortality [189].
CAR-T cells are ex vivo genetically engineered T cells programmed to induce a cytotoxic immune response
after
the recognition of specific tumor-associated antigens, such as CD19 in B-cell malignancies
(tisagenlecleucel and
axicabtagene ciloleucel) or BCMA in MM[144, 189]. Clinical trials of CAR-T cell therapy have reported a
low
incidence of cardiotoxicity, possibly due to patient enrollment criteria. However, subsequent
retrospective
cohort studies highlighted the occurrence of major adverse cardiovascular events (MACE) in 10-20% of
patients
[189]. These include tachyarrhythmia, hypotension, troponin elevation, cardiomyopathy, pericardial and
pleural
disorders, VTE and cardiogenic shock [190–192].
The exact mechanisms of T-cell therapy–induced cardiotoxicity remain elusive. The predominant mechanism
involves
indirect cardiotoxic effects following immune system activation: tumor-directed inflammation within the
microenvironment can lead to a systemic cytokine storm, known as cytokine release syndrome (CRS), that
impairs
cardiac function [193–195]. CRS is characterized by increased levels of pro-inflammatory cytokines,
primarily
IL-6, IL-1, TNF-α, and nitric oxide, released by activated T cells, macrophages and monocytes [196]. IL-6
has
been proposed to activate the gp130/STAT3 signaling pathway and induce oxidative stress. Subsequently,
this
leads to mitochondrial dysfunction, cardiomyocyte apoptosis and cardiac hypertrophy. In addition, IL-6
modifies
calcium handling and impairs myocardial contractility [197]. CRS treatment involves the administration of
tocilizumab, (an antibody binding and blocking the soluble and transmembrane IL-6 receptor), particularly
in
severe cases, to decrease the risk of cardiotoxicity [189]. Additional proposed mechanisms for CAR-T
cell–associated cardiotoxicity include the direct T cell-mediated damage to cardiac tissue due to
cross-reactivity with cardiac antigens via molecular mimicry or immune alloreactivity. Moreover, T cells
can
inadvertently recognize and attack heart-specific proteins unrelated to the tumor, leading to off-target
cardiac
toxicity [189].
BiTEs are fusion proteins with 2 different antigen-binding sites: one directed against the CD3 molecule,
which
leads to downstream activation of cytotoxic T lymphocytes, and another directed specifically at an antigen
present on malignant cells [198]. Blinatumomab, a bispecific CD19/CD3 antibody targeting CD19 on B cells,
has
been approved for R/R CD19-positive B-cell precursor ALL[199]. Data regarding the incidence of
cardiotoxicity
are limited, but in clinical practice, it has been associated with CRS, tachycardia and HF[189].
5.4 Intracellular targets on B cells
PIs form the cornerstone of combination regimens for patients with MM and AL amyloidosis, although they
are also
used for treating other malignancies. PIs target the chymotrypsin-activity of the 20S unit of the
proteasome
[200], causing proteome instability due to the accumulation of aggregated, unfolded, and/or damaged
polypeptides; this sustained proteome instability induces cell death.
Disruption of proteasome function has been associated with the development of CVD[201]. Accumulation of
misfolded proteins has been observed in various cardiomyopathies, including hypertrophic, dilated, and
desmin-related forms [202], and significant impairment of proteasomal activity has been documented in HF
and in
myocardial tissue obtained from patients with hypertrophic cardiomyopathy [203]. Therefore,
pharmacological
inhibition of the proteasome can be particularly harmful in cases with pre-existing cardiac dysfunction or
advanced age.
Carfilzomib, an intravenous irreversible PI, is associated with increased incidence of hypertension
(9-27%)[204,
205], HFrEF (4.1-16.2%)[204, 206], ischemic heart disease (1.8-17.6%), and arrhythmias (2.4-7%)[206, 207].
Bortezomib, an intravenous reversible PI, is also associated with CV toxicity, albeit at a lower degree
compared
to carfilzomib [147, 200]. The only orally administered PI ixazomib is generally not associated with a
high risk
for CV toxicity, apart from scarce evidence [200].
Bortezomib's cardiotoxicity is associated with impaired cardiomyocyte survival and contractility due to
protein
accumulation, mitochondrial dysfunction and worsening of the atherosclerotic plaque vulnerability [144,
208].
The higher risk of CV toxicity of carfilzomib is likely linked to irreversible inhibition of the
proteasome’s
proteolytic activity and the broader dosing spectrum [200]. The potential underlying mechanism of
cardiotoxicity
and LV dysfunction involves mitochondrial dysfunction [209]. In a cell model using human induced
pluripotent
stem cell-derived cardiomyocytes, carfilzomib reduced mitochondrial membrane potential, ATP production,
and
mitochondrial oxidative respiration, resulting in decreased cardiomyocyte contractility. Carfilzomib
treatment
also downregulated gene expression of extracellular matrices, the integrin complex, and cardiac
contraction
[144, 210]. Impaired autophagic signaling was observed with carfilzomib in vivo models of young and
aged
mice [211, 212]. Additionally, data indicate a possible role of the pyruvate oxidation pathway in
mitochondrial
dysfunction, as evidenced by the down-regulation of pyruvate and up-regulation of lactate dehydrogenase B
among
patients who experienced CV adverse events with carfilzomib [213]. Carfilzomib, like bortezomib,
affects
vascular smooth muscle cells, probably exacerbating the vulnerability of atherosclerotic plaques in
patients
[136].
Another important vascular complication observed with all proteasome inhibitors is thrombotic
microangiopathy
(TMA)[200, 214]. Proteasome inhibition can disrupt protein homeostasis within vascular cell walls,
promoting
cellular aging, cell cycle arrest, and programmed cell death [201]. Increasing in vitro evidence
supports
that the ubiquitin-proteasome pathway (UPP) regulates several endothelial cell functions, including the
expression and activation of endothelial nitric oxide synthase (eNOS), and the balance of vasodilatory and
vasoconstrictive signaling [215]. Additionally, the UPP contributes to vascular inflammation by modulating
NF-κB
activity and upregulating adhesion molecule expression [216]. While the full pathophysiologic mechanisms
are not
completely understood, microvascular injury appears to be linked, in part, to the suppression of vascular
endothelial growth factor (VEGF) signaling [200].
Paradoxically, under certain experimental conditions, short-term proteasome inhibition has shown
cardioprotective effects, particularly by attenuating or preventing left ventricular hypertrophy in animal
models of chronic pressure overload and in hypertensive Dahl salt-sensitive rats [217]. Moreover,
treatment with
proteasome inhibitors at low doses enhanced endothelial-dependent vasodilation in rat aortic rings in
vitro, via increasing the expression and activity of eNOS and reducing the levels of
endothelin-1[215,
218, 219].
Moreover, intracellular signal transduction downstream BCR, mediated by tyrosine kinases, mainly Bruton’s
tyrosine kinase (BTK) and PI3K, influences B-cell activation, proliferation and differentiation [220,
221].
Lately, BTK inhibitors and PI3K isoform-specific inhibitors have been approved for the treatment of
hematologic
malignancies.
Specifically, ibrutinib, an irreversible BTK inhibitor, has received approval for the treatment of CLL,
MCL, and
Waldenström’s macroglobulinemia. In the clinical setting, the drug is reported to induce cardiotoxicity,
mainly
arrhythmias and hypertension [220], possibly due to interactions with PI3K and other TEC pathways in
cardiomyocytes [144, 222]. In platelets, BTK inhibition is associated with central nervous system bleeding
or
ischemia, because it affects platelets’ activation [223, 224]. Recent evidence indicates that newer BTK
inhibitors, such as acalabrutinib and zanubrutinib, have increased selectivity for BTK, resulting in
reduced
cardiac implications [225, 226].
To date, three PI3K inhibitors have been approved for the treatment of CLL and indolent NHL: idelalisib,
copanlisib and duvelisib [221], but CV adverse events can occur, including arrhythmias and cardiac
dysfunction
due to the regulatory role of specific PI3K isoforms in the heart [129, 227].
Finally, Breakpoint cluster region (BCR)-Abelson (ABL) tyrosine kinase, is implicated in signal
transduction
pathways that regulate survival and proliferation in hematopoietic cells. It is used for the treatment of
CLL in
combination with other agents, such as imatinib, bosutinib and nilotinib [228, 229]. Inhibitors of BCR/ABL
cause
various forms of cardiotoxicity, such as congestive HF, AMI, coronary and peripheral artery disease,
peripheral
arterial occlusive disease, VTEs, and arrhythmias [129, 230]. The mechanism underlying these effects is
not
fully understood, but it appears to involve mitochondrial dysfunction [129, 231], autophagy and
cardiomyocyte
apoptosis [232, 233].
Taken together, B-cell–targeted immunomodulatory therapies have revolutionized the treatment of
hematologic
malignancies and autoimmune diseases, being increasingly recognized for their CV effects. Anti-CD20
therapy
(rituximab) has demonstrated potential cardioprotective effects by attenuating adverse remodeling in
preclinical
models and is under investigation in post-AMI settings in humans. CD38-targeting antibodies, such as
daratumumab
and isatuximab, can restore metabolic disequilibrium and calcium signaling by regulating NAD⁺ homeostasis,
though further mechanistic studies are needed. Other agents, including inotuzumab (anti-CD22) and
abatacept
(CTLA-4-Ig), show promising preclinical cardioprotective effects. On the contrary, ICIs can induce rare
but
severe immune-related cardiovascular toxicities, including myocarditis. Cellular therapies (CAR-T, BiTEs)
carry
risk for cytokine release syndrome–mediated cardiotoxicity, underscoring the need for careful monitoring.
Intracellular B-cell signaling inhibitors (BTK, PI3K, BCR-ABL inhibitors). PIs are associated with
arrhythmias,
hypertension, HF, and vascular complications, possibly due to mitochondrial and endothelial dysfunction
and
impaired autophagy. Collectively, these therapies highlight the dual potential for cardiovascular harm and
benefit, underscoring that targeting specific surface markers is more beneficial for the CV system than
tackling
intracellular pathways in B cells.
6. Conclusion
Despite decades of research, the multifaceted role of B cells —as antigen-presenting cells, cytokine producers, and antibody-secreting effectors— remains underrecognized as a driver of disease and a therapeutic target in CV pathology. Therapeutic interventions targeting intracellular signaling pathways in B cells are frequently associated with cardiotoxicity and CV adverse events. This is largely due to the shared signaling mechanisms —such as proteasome function and PI3K signaling— between B cells and cell types of the myocardium including cardiomyocytes and endothelial cells. Consequently, using such broad-spectrum immunomodulators such as proteasome inhibitors are likely unsuitable as therapeutic agents in CVD. Emerging therapies targeting intracellular pathways in B cells must be rigorously monitored in early clinical trials for CV adverse events.
In a translational perspective, testing B cell-related therapeutics for CVD with antibodies to target specific cell surface markers would be more rational. Targeting cell surface markers that are expressed by B cells such as CD20, CD38, or CD80, and modulating the BAFF pathway, represent a promising therapeutic approach, though repurposing existing agents for AMI or HF requires optimized, tailored dosing regimens. Future directions could involve the development of small molecule inhibitors of CD’s and BCMA or RNA silencing therapies to exploit B cell depletion and long-term cardioprotection, and to avoid the short-term antibody-related cardiotoxicity and CRS. Yet, the efficacy of these strategies is doubtful in hematologic malignancies.
Receptor specificity must be carefully balanced to preserve the protective functions of specific subsets such as B1 cells and Bregs [234] especially in the heart and avoid compromising overall immune competence. The broader concept of immunomodulation through metabolic reprogramming could serve as an alternative to modify HF outcomes [235–237]. More comprehensive studies are needed to better understand the contribution of each B cell subset in cardiac disease and hematologic malignancies, having as ultimate goal to optimize B cell-targeted therapies. In this context, a precision medicine approach should be adopted, integrating both disease characteristics and patient-specific immune profiles—particularly B-cell subset composition—into clinical decision-making. Standardized assays for circulating B-cell subtypes and antibody signatures should be validated in large, multicenter cohorts and incorporated into risk prediction models to enable early CVD risk stratification, therapeutic guidance, and monitoring of treatment response.
atherosclerosis, cardiac dysfunction, plaque instability, TMA, and platelet dysfunction. CRS (green shading), typically observed with CAR-T therapies and BiTEs, indirectly affects the cardiovascular system, while some therapies show no clear cardiovascular adverse events, or their impact remains uncertain (grey shading). By organizing drugs according to their molecular targets and linking them to cardiovascular outcomes, the figure underscores the dual role of B-cell–targeted therapies as both potential mediators of cardioprotection and contributors to cardiotoxicity.
CV: Cardiovascular; CRS: Cytokine release syndrome; HF: Heart failure; AEs: Adverse events; irAEs: immune-related adverse events; LV: Left ventricular; TMA: Thrombotic microangiopathy; CD: Cluster of differentiation; BAFF(-R): B-cell activating factor (Receptor); BCMA: B-cell maturation antigen; TACI: transmembrane activator and calcium-modulator and cyclophilin ligand interactor; APRIL: proliferation-inducing ligand; PD-1: programmed cell death protein- 1; PD-L1: programmed cell death ligand- 1; CTLA-4: cytotoxic T lymphocyte-associated antigen 4; BTK: Bruton’s tyrosine kinase; PI3K: Phosphatidylinositol-3-Kinase; BCR/ABL: Breakpoint cluster region/Abelson tyrosine kinase
Abbreviations
AL (Light chain-Amyloidosis); ALL (Acute lymphoblastic leukemia); ADCC (,Antibody-dependent cell-mediated cytotoxicity); AMI (Acute myocardial infarction); APRIL (Proliferation-inducing ligand); ARVC (Arrhythmogenic right ventricular cardiomyopathy); BAFF(-R) B-cell (activating factor (Receptor)); BiTEs (Bispecific T-cell engagers); BCMA (B-cell maturation antigen); BCR (B cell receptor); BCR/ABL (Breakpoint cluster region/Abelson tyrosine kinase); Bregs (B regulatory cells); BTK (Bruton’s tyrosine kinase); CAR-T (Chimeric Antigen Receptor T-cell); CCL (C-C chemokine ligand); CCR (C-C chemokine receptor); CD (Cluster of differentiation); CLL (Chronic lymphocytic leukemia); CLP (,Common lymphoid progenitors); CMP (,Common myeloid progenitors); CRS (Cytokine release syndrome); CSF1 (Macrophage colony-stimulating factor); CTLA-4 (Cytotoxic T lymphocyte-associated antigen 4); CV (Cardiovascular); CVD (Cardiovascular disease); CXCL ( ,CXC chemokine ligand); CXCR (CXC chemokine receptor); DCs (Dendritic cells); DCM (Dilated cardiomyopathy); DLBCL (Diffuse large B-cell lymphoma); D-VCd (Daratumumab, bortezomib, cyclophosphamide, and dexamethasone); eNOS (,Endothelial nitric oxide synthase); HC (Heavy chain); HL (Hodgkin lymphoma); FcγRs (Fc gamma receptors); (FL,Follicular lymphoma); HF (Heart failure); HFpEF (Heart failure with preserved ejection fraction); HFrEF (Heart failure with reduced ejection fraction); HSCs (,Hematopoietic Stem Cells); ICs (Immune checkpoints); ICIs (Immune checkpoint inhibitors); IFN-γ (Interferon gamma); Ig (Immunoglobulin); IgAP (Immunoglobulin-associated proteome); IL (Interleukin); IMiDs (Immunomodulatory drugs); IR (Ischemia-reperfusion); IRA (Innate response activator); irAEs (Immune-related adverse events); Kd (Carfilzomib and dexamethasone); KdD (Daratumumab, carfilzomib, and dexamethasone); LAG-3 (Lymphocyte-activation gene 3); LC (Light chain); LPS (Lipopolysaccharide); LVEF (,Left ventricular ejection fraction); GM-CSF (Granulocyte-macrophage colony-stimulating factor); mAb (Monoclonal antibodies); MACE (Major adverse cardiovascular events); mcMMAF (Maleimidocaproyl monomethylauristatin F); MZ (Marginal Zone); MG (Monoclonal gammopathy); MGTS (Monoclonal gammopathy of thrombotic significance); MGUS (Monoclonal gammopathy of undetermined significance); MHC (Major histocompatibility complex); MM (Multiple myeloma); MMF (Mycophenolate mofetil); NHE1 (Sodium-hydrogen exchanger-1); NFATC2 (Nuclear factor of activated T cells, cytoplasmic differentiation 2); NHL (Non-Hodgkin lymphoma); OxLDL (Oxidized low-density lipoprotein); PBMCs (Peripheral blood mononuclear cells); PD-1 (Programmed cell death protein-1); PD-L1 (Programmed cell death- ligand 1); PIs (Proteasome inhibitors); RA (Rheumatoid arthritis); R/R (Relapsed/Refractory); SDF1 (Stromal cell-derived factor 1); SGLT-2 (Sodium-glucose co-transporter-2); SLE (Systemic lupus erythematosus); TAC (Transverse aortic constriction); TACI (Transmembrane activator and calcium-modulator and cyclophilin ligand interactor); TGF (Transforming growth factor); TIGIT (Tumor-infiltrating T cell immunoreceptor with immunoglobulin and ITIM domain); TIM-1 (,T cell immunoglobulin and mucin domain-containing protein); TLRs (Toll-like receptors); TMA (Thrombotic microangiopathy); TNF-α (Tumor necrosis factor alpha); Tregs (T regulatory cells); UPP (Ubiquitin-proteasome pathway); VEGF (Vascular endothelial growth factor); VTE (Venous thromboembolism); WM (Waldenström’s macroglobulinemia);
Acknowledgements
AC and PEN were supported by the 2nd Call for Hellenic Foundation for Research and Innovation (H.F.R.I.) Research Projects to Support Faculty Members & Researchers “ElucidatioN of LIGHt chain amyloidosis induced cardioToxicity: EstablishMENT of in vitro and in vivo models” (ENLIGHTEnMENT) and devote this work to late Professor Ioanna Andreadou.
Disclosure Statement
DF: honoraria; Janssen, GSK, Sanofi; EK: honoraria and research support; Johnson & Johnson, Pfizer, GSK.
References
| 1 | Thakur S, Chandra A, Kumar V, Bharti S. Environmental pollutants: endocrine
disruptors/pesticides/reactive dyes and inorganic toxic compounds, metals, radionuclides, and
metalloids and their impact on the ecosystem. In Biotechnology for environmental sustainability,
2025 Feb 11 (pp. 55-100). Singapore: Springer Nature Singapore.
https://doi.org/10.1007/978-981-97-7221-6_3 |
| 2 | Isaac RA, Subbarayalu R, Kumar MS, Martin TM, Lo SC, Santosh W. Human endocrine disruption: an
updated review of toxicological insights into parabens and phthalates. Toxicology and Environmental
Health Sciences. 2025 May 29:1-4.
|
| 3 | Barabasz W, Pikulicka A, Wzorek Z, Nowak AK. Ecotoxicological aspects of the use of parabens in
the production of cosmetics. Technical Transactions. 2019 Dec 20;2019(Year 2019 (116)):99-124.
https://doi.org/10.4467/2353737XCT.19.126.11451 |
| 4 | Kim MJ, Kwack SJ, Lim SK, Kim YJ, Roh TH, Choi SM, Kim HS, Lee BM. Toxicological evaluation of
isopropylparaben and isobutylparaben mixture in Sprague- Dawley rats following 28 days of dermal
exposure. Regulatory Toxicology and Pharmacology. 2015 Nov 1;73(2):544-51.
https://doi.org/10.1016/j.yrtph.2015.08.005 |
| 5 | Safarova IR. Hydroxybenzoic acid derivatives and their biological activity. Processes of
Petrochemistry and Oil Refining. 2022;23(1).
|
| 6 | Liang J, Liu QS, Ren Z, Min K, Yang X, Hao F, Zhang Q, Liu Q, Zhou Q, Jiang G. Studying
paraben-induced estrogen receptor-and steroid hormone-related endocrine disruption effects via
multi-level approaches. Science of The Total Environment. 2023 Apr 15;869:161793.
https://doi.org/10.1016/j.scitotenv.2023.161793 |
| 7 | Amir S, Shah ST, Mamoulakis C, Docea AO, Kalantzi OI, Zachariou A, Calina D, Carvalho F, Sofikitis
N, Makrigiannakis A, Tsatsakis A. Endocrine disruptors acting on estrogen and androgen pathways
cause reproductive disorders through multiple mechanisms: a review. International journal of
environmental research and public health. 2021 Feb;18(4):1464.
https://doi.org/10.3390/ijerph18041464 |
| 8 | Kuru BB, Kuru M. Reproductive behavior in female rats: Reproductive behavior in female rats. Rats.
2023 Jul 13;1(1):21-6.
|
| 9 | Zhao M, Li Y, Wang Z. Mercury and mercury-containing preparations: history of use, clinical
applications, pharmacology, toxicology, and pharmacokinetics in traditional Chinese medicine.
Frontiers in Pharmacology. 2022 Mar 2;13:807807.
https://doi.org/10.3389/fphar.2022.807807 |
| 10 | Krout IN. The Role of Methylmercury Demethylation in Modulating Human Toxicokinetic Outcomes.
University of Rochester; 2023.
|
| 11 | Rupa SA, Patwary MA, Matin MM, Ghann WE, Uddin J, Kazi M. Interaction of mercury species with
proteins: towards a possible mechanism of mercurial toxicology. Toxicology research. 2023
Jun;12(3):355-68.
https://doi.org/10.1093/toxres/tfad039 |
| 12 | Azevedo LF, Karpova N, Rocha BA, Barbosa Junior F, Gobe GC, Hornos Carneiro MF. Evidence on
neurotoxicity after intrauterine and childhood exposure to organomercurials. International Journal
of Environmental Research and Public Health. 2023 Jan 7;20(2):1070.
https://doi.org/10.3390/ijerph20021070 |
| 13 | Sisk C, Lonstein JS, Gore AC. Critical periods during development: Hormonal influences on
neurobehavioral transitions across the life span. In Neuroscience in the 21st century 2013 (pp.
1715-1752). Springer, New York, NY.
https://doi.org/10.1007/978-1-4614-1997-6_61 |
| 14 | Grandjean P, Barouki R, Bellinger DC, Casteleyn L, Chadwick LH, Cordier S, Etzel RA, Gray KA, Ha
EH, Junien C, Karagas M. Life-long implications of developmental exposure to environmental
stressors: new perspectives. Endocrinology. 2015 Oct 1;156(10):3408-15.
https://doi.org/10.1210/en.2015-1350 |
| 15 | Vrolyk V, Tremblay C, Picut CA. Juvenile Toxicology. In Drug Discovery and Evaluation: Safety and
Pharmacokinetic Assays 2024 Oct 22 (pp. 2337-2371). Cham: Springer International Publishing.
https://doi.org/10.1007/978-3-031-35529-5_121 |
| 16 | Srivastava AK, Negi G. Role of nonclinical programs in drug development. In The Quintessence of
Basic and Clinical Research and Scientific Publishing 2023 Oct 1 (pp.579-593). Singapore: Springer
Nature Singapore.
https://doi.org/10.1007/978-981-99-1284-1_35 |
| 17 | Sewell F, Lewis D, Mehta J, Terry C, Kimber I. Rethinking agrochemical safety assessment: A
perspective. Regulatory Toxicology and Pharmacology. 2021 Dec 1;127:105068.
https://doi.org/10.1016/j.yrtph.2021.105068 |
| 18 | Manservisi F. Reproductive and developmental toxicity study using Sprague-Dawley rats exposed
under various calendars to the weedkiller Glyphosate and commercial formulations Glyphosate-based.
2021.
|
| 19 | Malashetty V, Deshpande R, Patil S. Seventy‐Day Toxicity Study in Juvenile SpragueDawley Rats with
Semicarbazide (SEM) from Weaning to Sexual Maturity. Journal of Toxicology. 2022;2022(1):5059761.
https://doi.org/10.1155/2022/5059761 |
| 20 | Sharma A. The role of puberty in shaping the ovarian reserve and reproductive lifespan (Doctoral
dissertation, University of Otago); 2024.
|
| 21 | Edwards PM, Parker VH. A simple, sensitive, and objective method for early assessment of
acrylamide neuropathy in rats. Toxicology and applied pharmacology.1977 Jun 1;40(3):589-91.
https://doi.org/10.1016/0041-008X(77)90083-7 |
| 22 | Han B, Lv Z, Han X, Li S, Han B, Yang Q, Wang X, Wu P, Li J, Deng N, Zhang Z. Harmful effects of
inorganic mercury exposure on kidney cells: mitochondrial dynamics disorder and excessive oxidative
stress. Biological Trace Element Research. 2022 Apr;200(4):1591-7.
https://doi.org/10.1007/s12011-021-02766-3 |
| 23 | Rivera-Núñez Z, Kinkade CW, Zhang Y, Rockson A, Bandera EV, Llanos AA, Barrett ES. Phenols,
parabens, phthalates and puberty: a systematic review of synthetic chemicals commonly found in
personal care products and girls' pubertal development. Current environmental health reports. 2022
Dec;9(4):517-34.
https://doi.org/10.1007/s40572-022-00366-4 |
| 24 | Odenbro A. Effects on reproduction and hormone metabolism. In Biological Effects of Organolead
Compounds 2020 Mar 25 (pp. 161-175). CRC Press.
https://doi.org/10.1201/9780429282980-13 |
| 25 | Akmal H, Ahmad S, Abbasi MH, Jabeen F, Shahzad K. A study on assessing the toxic effects of ethyl
paraben on rohu (Labeo rohita) using different biomarkers; hematobiochemical assays, histology,
oxidant and antioxidant activity, and genotoxicity. PLOS ONE. 2024 May 6;19(5):e0302691.
https://doi.org/10.1371/journal.pone.0302691 |
| 26 | Nicholson JK, Timbrell JA, Sadler PJ. Proton NMR spectra of urine as indicators of renal damage.
Mercury-induced nephrotoxicity in rats. Molecular Pharmacology. 1985 Jun 1;27(6):644-51.
https://doi.org/10.1016/S0026-895X(25)12559-5 |
| 27 | Parent AS, Teilmann G, Juul A, Skakkebaek NE, Toppari J, Bourguignon JP. The timing of normal
puberty and the age limits of sexual precocity: variations around the world, secular trends, and
changes after migration. Endocrine Reviews. 2003 Oct;24(5):668693.
https://doi.org/10.1210/er.2002-0019 |
| 28 | Ye X, Pan W, Zhao S, Zhao Y, Zhu Y, Liu J, Liu W. Relationships of pyrethroid exposure with
gonadotropin levels and pubertal development in Chinese boys. Environmental Science & Technology.
2017 Jun;51(11):6379-6386.
https://doi.org/10.1021/acs.est.6b05984 |
| 29 | Yesumanipreethi S, Nirmal Magadalenal N, Moses Inbaraj R. Impact of phthalates and parabens on the
neurobehavioral and reproductive function: a review. InProceedings of the Zoological Society 2021
Dec (Vol. 74, No. 4, pp. 572-590). New Delhi: Springer India.
https://doi.org/10.1007/s12595-021-00408-z |
| 30 | Lee JD, Bae JS, Kim HY, Song SW, Kim JC, Lee BM, Kim KB. Repeated-dose toxicity and toxicokinetic
study of isobutylparaben in rats subcutaneously treated for 13 weeks. Archives of Toxicology. 2024
Jul;98(7):2231-46.
https://doi.org/10.1007/s00204-024-03741-2 |
| 31 | Panghal A, Jena G. Single versus intermittent cycle exposure effect of 6mercaptopurine in juvenile
Sprague-Dawley rat: a germ cell-specific mechanistic study. Naunyn-Schmiedeberg's Archives of
Pharmacology. 2024 May;397(5):3155-68.
https://doi.org/10.1007/s00210-023-02797-8 |
| 32 | Ullah H, Khan MF, Hashmat F. In vitro, concentration and time-dependent effect of phenyl mercuric
acetate on the chemical status of glutathione (GSH). Gomal University Journal of Research. 2012 Dec
31;28(2):1-8.
|